
by codm | May 24, 2023 | Newsletter
For Print (PDF)
Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) announced today the presentation of research across various types of cancer from its oncology portfolio and pipeline during the 2023 American Society of Clinical Oncology (ASCO) Annual Meeting (#ASCO23), which is taking place virtually and in-person in Chicago, Illinois from June 2 to 6.
Notable research includes an oral presentation of results from the final pre-specified overall survival analysis of the pivotal Phase 3 CLEAR (Study 307)/KEYNOTE-581 trial, which evaluated lenvatinib (LENVIMA®) plus pembrolizumab (KEYTRUDA®) versus sunitinib for the first-line treatment of patients with advanced renal cell carcinoma (Abstract #4502). A post hoc analysis from the REFLECT trial evaluating lenvatinib monotherapy versus sorafenib in the first-line treatment of patients with unresectable hepatocellular carcinoma (HCC) will also be shared in a poster presentation (Abstract #4078).
“The outlook for advanced renal cell carcinoma has evolved in recent years, and the final analysis from the pivotal CLEAR trial to be presented at ASCO represents another step forward for patients and an opportunity to provide their physicians with long-term data,” said Dr. Takashi Owa, Chief Scientific Officer, Senior Vice President, Eisai Co., Ltd. “New data for lenvatinib and from our oncology pipeline showcase Eisai’s continued commitment to driving innovation and exploring novel therapeutic modalities in our ambition to live out our human health care concept, our corporate mission to meet the needs of more people who face a cancer diagnosis.”
Additional data from Eisai’s pipeline include a poster presentation of findings from a phase 1b study of E7386, a CREB-binding protein (CBP) / β-catenin interaction inhibitor, in combination with lenvatinib in patients with advanced HCC (Abstract #4075), and the small cell lung cancer cohort of a phase 1b/2 trial evaluating E7389-LF, a new liposomal formulation of eribulin, in combination with nivolumab (Abstract #8593). Insights from preclinical testing of farletuzumab ecteribulin (FZEC), formerly known as MORAb-202, and MORAb-109, antibody drug conjugates (ADC), in rare gynecologic cancers will also be published online (Abstract # e17634).
Furthermore, Bliss Biopharmaceutical Co., Ltd. (BlissBio) will present a poster at the conference with results from the first-in-human study of BB-1701, a HER2-targeting ADC (Abstract #3029). Eisai entered into a joint development agreement with BlissBio for BB-1701 with option rights for a strategic collaboration in April 2023. A Phase 1/2 clinical study of BB-1701 in the U.S. and China for HER2-expressing solid tumors is currently underway.
This release discusses investigational compounds and investigational uses for FDA-approved products. It is not intended to convey conclusions about efficacy and safety. There is no guarantee that any investigational compounds or investigational uses of FDA-approved products will successfully complete clinical development or gain FDA approval.
The full list of presentations is included below. These abstracts will be made available on Thursday, May 25, 2023 at 4:00 PM Central Daylight Time (CDT).
| Cancer Type |
Study/Compound |
Abstract Title |
Abstract Type & Details |
| Lenvatinib Plus Pembrolizumab |
| Gynecologic Cancer |
CLEAR |
Final prespecified overall survival (OS) analysis of CLEAR: 4-year follow-up of lenvatinib plus pembrolizumab (L+P) vs sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC) |
Oral Abstract Session
Abstract #4502
June 5, 2023
11:54 AM CDT
|
|
Lenvatinib
|
| Gastrointestinal Cancer |
REFLECT |
Efficacy of lenvatinib (LEN) vs sorafenib (SOR) in the first-line (1L) treatment of patients (pts) with unresectable hepatocellular carcinoma (uHCC): A post hoc analysis of patients with nonviral etiology from REFLECT |
Poster Session
Abstract #4078
June 5, 2023
8:00 AM CDT
|
| Pipeline |
| Lung Cancer |
E7389-LF |
Phase 2 small cell lung cancer (SCLC) cohort of a phase 1b/2 trial of a liposomal formulation of eribulin in combination with nivolumab |
Poster Session
Abstract #8593
June 4, 2023
8:00 AM CDT
|
| Gastrointestinal Cancers |
E7386 (plus lenvatinib) |
A phase 1b study of E7386, a CREB-binding protein (CBP)/β-catenin interaction inhibitor, in combination with lenvatinib in patients with advanced hepatocellular carcinoma |
Poster Session
Abstract #4075
June 5, 2023
8:00 AM CDT
|
| Gynecologic Cancer |
Farletuzumab Ecteribulin
(FZEC) |
Preclinical testing of farletuzumab ecteribulin (FZEC [MORAb-202]) and MORAb-109, folate receptor α and mesothelin targeting antibody-drug conjugates (ADCs), in rare gynecologic cancers |
Online Publication
Abstract #e17634
May 25, 2023
4:00 PM CDT
|
| Solid tumors |
BB-1701
(Presented by BlissBio) |
A first-in-human, open label, multiple dose, dose escalation, and cohort expansion phase 1 study to investigate the safety, tolerability, pharmacokinetics and antitumor activity of BB-1701 in patients with locally advanced/metastatic HER2-expressing solid tumors |
Poster Session
Abstract #3029
June 3, 2023
8:00 AM CDT
|
| Additional Research |
| Pan-tumor |
Systematic review |
Anti-drug antibodies related to CTLA-4, PD-1 or PD-L1 inhibitors across tumour types: A systematic review |
Online Publication
Abstract #e14600
May 25, 2023
4:00 PM CDT
|
In March 2018, Eisai and Merck & Co., Inc., Rahway, NJ, USA (known as MSD outside the United States and Canada), through an affiliate, entered into a strategic collaboration for the worldwide co-development and co-commercialization of lenvatinib, both as monotherapy and in combination with Merck’s anti-PD-1 therapy pembrolizumab. Eisai and Merck are studying the LENVIMA plus KEYTRUDA combination through the LEAP (LEnvatinib And Pembrolizumab) clinical program in various tumor types across more than multiple clinical trials.
In June 2021, Eisai and Bristol Myers Squibb entered into an exclusive global strategic collaboration agreement for the co-development and co-commercialization of farletuzumab ecteribulin (FZEC, formerly known as MORAb-202), a folate receptor alpha (FRα)-targeting ADC. Eisai and Bristol Myers Squibb are currently investigating FZEC in multiple studies including: a Phase 1/2 clinical study in the United States and Europe for solid tumors including endometrial cancer, a Phase 2 clinical study in the United States and Europe for non-small cell lung cancer, and a Phase 2 clinical study in Japan, the United States and Europe for ovarian cancer, peritoneal cancer and fallopian tube cancer.
Media Inquiries:
Public Relations Department,
Eisai Co., Ltd.
+81-(0)3-3817-5120
[Notes to editors]
1. Eisai’s Focus on Cancer
Eisai acknowledges “Oncology” as one of its key strategic areas, and will continue to focus on the discovery and development of anti-cancer drugs within drug discovery domains including “tumor microenvironment”, “proteostasis disruption”, “cell linage and cell differentiation”, and “inflammation, hypoxia, oxidative stress and cell senescence” under the Deep Human Biology Learning (DHBL) drug discovery and development organization. Eisai aspires to discover innovative new drugs with new targets and mechanisms of action from these domains with the aim of contributing to the cure of cancers.
* KEYTRUDA® is a registered trademark of Merck Sharp & Dohme LLC., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
by codm | May 22, 2023 | Newsletter
For Print (PDF)
INVESTOR, PHARMACEUTICAL AND TRADE MEDIA INFORMATION ONLY. THIS PRESS RELEASE IS NOT FOR A UK AUDIENCE.
TOKYO and CAMBRIDGE, Mass., May 22, 2023 – Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, “Biogen”) announced today that Eisai has submitted a Marketing Authorization Application (MAA) for lecanemab, an investigational anti-amyloid beta (Aβ) protofibril antibody, for the treatment of early Alzheimer’s disease (mild cognitive impairment due to Alzheimer’s disease (AD) and mild AD dementia) with confirmed amyloid pathology in the brain, to the UK Medicines and Healthcare products Regulatory Agency (MHRA) in Great Britain. Lecanemab has been designated by the MHRA for the Innovative Licensing and Access Pathway (ILAP).
The MAA is based on the results of the confirmatory Phase III Clarity AD study and Phase IIb clinical study (Study 201), which demonstrated that lecanemab treatment showed a reduction of clinical decline in early AD, and is subject to a validation to determine whether it will be accepted by the MHRA. Lecanemab selectively binds and eliminates soluble, toxic Aβ aggregates (protofibrils) that are thought to contribute to the neurotoxicity in AD. As such, lecanemab may have the potential to have an effect on disease pathology and the progression of the disease. The Clarity AD study of lecanemab met its primary endpoint and all key secondary endpoints with highly statistically significant results.
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
Contacts:
| Eisai |
Biogen Inc. |
MEDIA CONTACT:
Eisai Co., Ltd.
Public Relations Department
TEL: +81 (0)3-3817-5120
Eisai Inc. (U.S.)
Libby Holman
TEL: + 1-201-753-1945
Libby_Holman@eisai.com
Eisai Europe, Ltd.
(Europe, Australia, New Zealand and Russia)
EMEA Communications Department
TEL: +44-(0) 786 601 1272
EMEA-comms@eisai.net
INVESTOR CONTACT:
Eisai Co., Ltd.
Investor Relations Department
TEL: +81 (0) 3-3817-5122 |
MEDIA CONTACT:
Jack Cox
+ 1-210-544-7920
public.affairs@biogen.com
INVESTOR CONTACT:
Chuck Triano
+ 1-781-464-2442
IR@biogen.com |
[Notes to editors]
1. About the Innovative Licensing and Access Pathway (ILAP) in the UK
The ILAP is a program offered by the MHRA (UK) for development programs with the goal of reducing the time to market for innovative medicines that treat life-threatening or seriously debilitating conditions and/or conditions for which there is a significant unmet patient need. The ILAP aims to achieve this goal by enabling enhanced coordination between sponsors, the MHRA and reimbursement bodies such as National Institute for Health and Care Excellence (NICE), leading up to Marketing Authorization Application (MAA) submissions to support accelerated access.
2. About Lecanemab
Lecanemab is the result of a strategic research alliance between Eisai and BioArctic. It is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (protofibril) and insoluble forms of amyloid-beta (Aβ).
In the U.S., lecanemab was granted accelerated approval for Alzheimer’s disease (AD) by the U.S. Food and Drug Administration (FDA) on January 6, 2023. On the same day, Eisai submitted a supplemental Biologics License Application (sBLA) to the FDA for approval under the traditional pathway. This application was accepted, and has been granted Priority Review, with a Prescription Drug User Fee Act (PDUFA) action date of July 6, 2023. In Europe, Eisai submitted a MAA to the European Medicines Agency (EMA) on January 9, 2023, which was accepted on January 26, 2023. In Japan, Eisai submitted a Marketing Authorization Application to the Pharmaceuticals and Medical Devices Agency (PMDA) on January 16, 2023, and Priority Review was designated by the Ministry of Health, Labour and Welfare (MHLW) on January 26, 2023. In China, Eisai initiated submission of data for a BLA to the National Medical Products Administration (NMPA) of China in December 2022, which was designated for Priority Review on February 27, 2023. In Canada, Eisai submitted a New Drug Submission (NDS) to Health Canada on March 31, 2023, and was accepted on May 15 of the same year.
Lecanemab is indicated for the treatment of AD in the U.S. Treatment with lecanemab should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in Aβ plaques observed in patients treated with lecanemab. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
Eisai has completed a lecanemab subcutaneous bioavailability study, and subcutaneous dosing is currently being evaluated in the Clarity AD OLE.
Since July 2020 the Phase III clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen. The Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, has been ongoing since January 2022.
3. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
4. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
5. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), with working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter, LinkedIn and Facebook. For more information about Eisai in the EMEA region please visit www.eisai.eu.
6. About Biogen
Founded in 1978, Biogen is a leading global biotechnology company that has pioneered multiple breakthrough innovations including a broad portfolio of medicines to treat multiple sclerosis, the first approved treatment for spinal muscular atrophy, and two co-developed treatments to address a defining pathology of Alzheimer’s disease. Biogen is advancing a pipeline of potential novel therapies across neurology, neuropsychiatry, specialized immunology and rare diseases and remains acutely focused on its purpose of serving humanity through science while advancing a healthier, more sustainable and equitable world.
The company routinely posts information that may be important to investors on its website. Follow Biogen on social media – Twitter, LinkedIn, Facebook, YouTube.
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements speak only as of the date of this press release. We do not undertake any obligation to publicly update any forward-looking statements.
by codm | May 17, 2023 | Newsletter
For Print (PDF)
Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) today announced the publication of results from a simulation study evaluating the societal value of anti-amyloid-beta (Aβ) protofibril* antibody lecanemab (generic name, U.S. brand name: LEQEMBI™) in individuals living with mild cognitive impairment (MCI) due to Alzheimer’s disease (AD) and mild AD dementia (collectively known as early AD) in the context of the Japanese health care system in the peer-reviewed journal Neurology and Therapy. The paper concluded that lecanemab treatment would improve health and humanistic (quality of life) outcomes and reduce economic burden for individuals with early AD and their caregivers in Japan.
This model-based simulation was conducted using data from the Phase 3 Clarity AD study evaluating the efficacy and safety of lecanemab for early AD with confirmed amyloid pathology by applying an academically validated disease simulation model (AD Archimedes Condition Event simulation: AD ACE model1,2) as well as government statistics, including Japanese epidemiological data and fact-finding surveys on long-term care, and other past research articles to take into account the environment under the Japanese healthcare system from the healthcare payer’s perspective, focusing on direct care costs (including out- and in-patient services, nursing- and home-health- care services, cost of medications, and other intervention costs) and the societal perspective (social costs including informal care costs such as family nursing care in addition to direct care costs). In this paper, the cost reduction effect of lecanemab and the improvement in health outcomes were integrated, incorporating the former cost reduction effect as is, while the latter outcome improvement effect was estimated based on previous studies in the U.S. and the benchmark price estimation process of the U.S. Institute for Clinical and Economic Review (ICER).
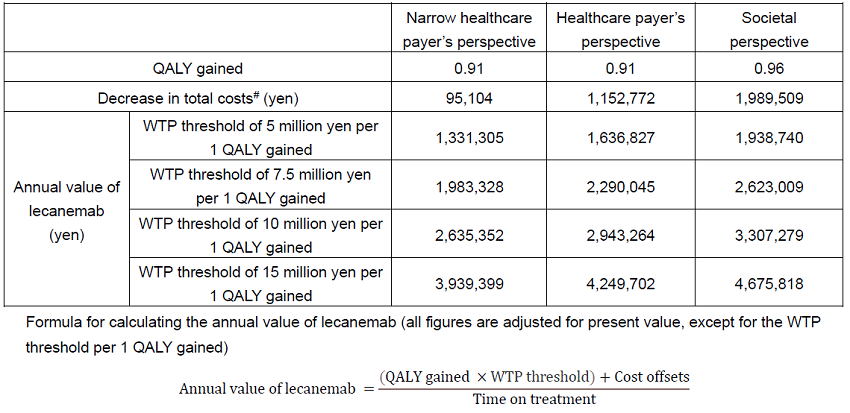
The analysis predicted an increase in Quality-Adjusted Life-Years** (QALYs) by 0.91 from both the narrow healthcare payer’s perspective (considering only medical treatment costs) and the healthcare payer’s perspective (medical treatment and public care costs) (SoC (standard of care***): 6.12, lecanemab+SoC (lecanemab treatment with SoC): 7.03). From the societal perspective, the increase in QALYs was estimated to be 0.96 (SoC: 5.78, lecanemab+SoC: 6.74) compared to SoC. In addition, compared to SoC alone, lecanemab+SoC was predicted to result in a decrease in total costs of 95,104 yen from the narrow healthcare payer’s perspective (SoC: 2,793,491 yen, lecanemab+SoC: 2,698,387: yen), 1,152,772 yen from the healthcare payer’s perspective (SoC: 11,381,044 yen, lecanemab+SoC: 10,228,272 yen), and 1,989,509 yen from the societal perspective (SoC: 24,482,321 yen, lecanemab+SoC 22,492,811 yen). The estimated mean duration of lecanemab treatment was 3.68 years in this simulation.
While taking into account the above factors, the model estimated that the annual value of lecanemab was 1,331,305 yen to 3,939,399 yen from the narrow healthcare payer’s perspective, 1,636,827 yen to 4,249,702 yen from the healthcare payer’s perspective, and 1,938,740 yen to 4,675,818 yen from the societal perspective at the willingness-to-pay (WTP) threshold of 5 million yen to 15 million yen, per 1 QALY gained under the Japanese healthcare system. Regarding the WTP threshold to be paid per 1 QALY gained, an academic article has proposed that 5 times per capita gross domestic product (GDP) is appropriate for severe AD,3 and from an international perspective, it is thought that for common diseases other than AD, the WTP threshold per 1 QALY gained is equivalent to 1-3 times per capita GDP. Considering the above, this paper evaluated that 15 million yen per 1 QALY is appropriate as the WTP threshold for assessing the societal value of lecanemab.
Since AD has a huge impact not only on medical costs but also public care costs as well as the invisible burden including family care, it is important to recognize the societal value, and this paper showed that annual value of lecanemab from the societal perspective can be up to 4,675,818 yen.
Health outcomes (QALYs) and decrease in total costs from lecanemab treatment, and annual value of lecanemab
“Approximately 50% of public long-term care insurance finances in Japan are spent on care costs due to AD (public long-term care insurance finances: 9,626.6 billion yen; public long-term care costs due to AD: 4,783.2 billion yen in 2018). It is understood that AD has a significant impact on not only medical costs but also long-term care insurance finance and the costs due to AD increases significantly with the progression of the pathological stage, and when comparing MCI and severe dementia, medical costs and long-term care costs increase significantly by about 2 times and 10 times, respectively.4 The simulation results show that, in Japan, treatment with lecanemab can have a significant impact on caregivers and society as a whole by improving delaying disease progression”, said Masatomi Akana, Senior Vice President, Chief Government Relations Officer, and Global Value & Access Eisai Co., Ltd., “We believe that this paper will be important information for stakeholders to better understand the potential clinical and socio-economic value of lecanemab in Japan, as well as for discussions on the evaluation of innovation by pharmaceutical companies. We will continue to release data and information transparently and promptly in order to deliver lecanemab to eligible early AD patients.”
“This paper followed the U.S. methodology, and added country-specific conditions to estimate the societal value of the medicine in Japan” said the first author of this paper, Dr. Ataru Igarashi, Associate Professor of Medicine at Yokohama City University and Visiting Associate Professor of Pharmaceutical Policy at the University of Tokyo Graduate School of Pharmaceutical Sciences. “When assessing the value of medicines, not only for dementia but for all diseases, I believe that in addition to efficacy, safety and treatment cost, various other aspects should be taken into account, such as the impact on work and the degree of burden reduction for family members and medical personnel.”
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
* Protofibrils are large Aβ aggregated soluble species of 75-5000 Kd.5
** The quality-adjusted life year (QALY) is a measure of the value of health outcomes. Since health is a function of length of life (i.e., quantity) and quality of life (QOL), the QALY was developed as an attempt to combine the value of these attributes into a single index number. One QALY equates to one year in perfect health. QOL scores range from 1 (full health) to 0 (dead). For example, if a new treatment and an existing treatment both increase survival years by 3 years, but the new treatment maintains a QOL of 0.7 (QALY=2.1), while the existing treatment has a lower QOL of 0.5 (QALY=1.5), the incremental QALY for the new treatment would be 0.6 (QALY = QOL score x survival years).
*** Standard of Care (SoC) for AD currently consists of lifestyle modifications and pharmacologic treatment of symptoms.
1 Tahami Monfared AA, Tafazzoli A, Ye W, Chavan A, Zhang Q. Long-Term Health Outcomes of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling. Neurology and therapy. 2022;11(2):863-80.
2 Tahami Monfared AA, Tafazzoli A, Chavan A, Ye W, Zhang Q. The Potential Economic Value of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling. Neurology and Therapy. 2022;11(3):1285-307.
3 Lakdawalla DN, Phelps CE. Health technology assessment with risk aversion in health. J Health Econ. 2020;72:102346. doi: 10.1016/j.jhealeco.2020.102346
4 S. Ikeda, M. Mimura, M. Ikeda, K .Wada-Isoe, M. Azuma, S. Inoue, K. Tomita. Economic Burden of Alzheimer’s Disease Dementia in Japan. Journal of Alzheimer’s Disease 81(2021)309-319
5 Söderberg, L., Johannesson, M., Nygren, P. et al. Lecanemab, Aducanumab, and Gantenerumab — Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics (2022).
Media Inquiries:
Public Relations Department,
Eisai Co., Ltd.
+81-(0)3-3817-5120
Eisai Inc (U.S.)
Libby Holman
201-753-1945
Libby_Holman@eisai.com
[Notes to editors]
1. About Lecanemab
Lecanemab (brand name in the U.S.: LEQEMBI™) is the result of a strategic research alliance between Eisai and BioArctic. Lecanemab is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (protofibril) and insoluble forms of amyloid-beta (Aβ). In the U.S., LEQEMBI was granted accelerated approval by the U.S. Food and Drug Administration (FDA) on January 6, 2023. LEQEMBI is indicated for the treatment of Alzheimer’s disease (AD) in the U.S. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved in the U.S. under Accelerated Approval based on reduction in Aβ plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
Please see full Prescribing Information in the United States.
In the U.S., Eisai submitted a supplemental Biologics License Application (sBLA) to the FDA for approval under the traditional pathway on January 6, 2023. On March 3, 2023, the FDA accepted Eisai’s sBLA based on the Clarity AD clinical data, and the LEQEMBI application has been granted Priority Review, with a Prescription Drug User Fee Act (PDUFA) action date of July 6, 2023. The FDA is planning to hold an Advisory Committee to discuss this application on June 9, 2023. Eisai submitted an application for manufacturing and marketing approval to the Pharmaceuticals and Medical Devices Agency (PMDA) on January 16, 2023, in Japan. The Priority Review was granted by the Ministry of Health, Labour and Welfare (MHLW) on January 26, 2023. Eisai utilized the prior assessment consultation system of PMDA, with the aim of shortening the review period for lecanemab. In Europe, Eisai submitted a marketing authorization application (MAA) to the European Medicines Agency (EMA) on January 9, 2023, and accepted on January 26, 2023. In China, Eisai initiated submission of data for a BLA to the National Medical Products Administration (NMPA) of China in December 2022, and the Priority Review was granted on February 27, 2023. In Canada, Eisai submitted a New Drug Submission (NDS) to Health Canada on March 31, 2023, and was accepted on May 15 of the same year.
Eisai has completed lecanemab subcutaneous bioavailability study, and subcutaneous dosing is currently being evaluated in the Clarity AD (Study 301) OLE.
Since July 2020 the Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing.
2. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
3. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
by codm | May 16, 2023 | Newsletter
For Print (PDF)
TOKYO and CAMBRIDGE, Mass., May 16, 2023 – Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, “Biogen”) announced today that Health Canada has accepted a New Drug Submission (NDS) for lecanemab (brand name in the U.S.: LEQEMBI™), an investigational anti-amyloid beta (Aβ) protofibril* antibody, for the treatment of early Alzheimer’s disease (mild cognitive impairment due to Alzheimer’s disease (AD) and mild AD dementia) with confirmed amyloid pathology in the brain.
The NDS is based on the results of the Phase III Clarity AD study and Phase IIb clinical study (Study 201), which demonstrated the lecanemab treatment showed a reduction of clinical decline in early AD. Lecanemab selectively binds and eliminates soluble, toxic Aβ aggregates (protofibrils) that are thought to contribute to the neurotoxicity in AD. As such, lecanemab may have the potential to have an effect on disease pathology and to slow down the progression of the disease. The Clarity AD study of lecanemab met its primary endpoint and all key secondary endpoints with highly statistically significant results. In November 2022, the results of the Clarity AD study were presented at the 2022 Clinical Trials on Alzheimer’s Disease (CTAD) conference, and simultaneously published in the New England Journal of Medicine, a peer-reviewed medical journal.
Lecanemab was approved under the accelerated approval pathway in the U.S. and was launched in the U.S. on January 18, 2023. The accelerated approval was based on Phase II data that demonstrated that lecanemab reduced the accumulation of Aβ plaque in the brain, a defining feature of AD, and its continued approval may be contingent upon verification of lecanemab’s clinical benefit in a confirmatory trial. The U.S. Food and Drug Administration (FDA) determined that the results of Clarity AD can serve as the confirmatory study to verify the clinical benefit of lecanemab.
In the U.S., Eisai submitted a supplemental Biologics License Application (sBLA) to the FDA for approval under the traditional pathway on January 6, 2023. On March 3, 2023, the FDA accepted Eisai’s sBLA based on the Clarity AD clinical data, and the lecanemab application has been granted Priority Review, with a Prescription Drug User Fee Act (PDUFA) action date of July 6, 2023. The FDA is planning to hold an Advisory Committee to discuss this application on June 9, 2023. In Japan, Eisai submitted an application for manufacturing and marketing approval to the Pharmaceuticals and Medical Devices Agency (PMDA) on January 16, 2023. Priority Review was granted by the Ministry of Health, Labour and Welfare (MHLW) on January 26, 2023. Eisai utilized the PMDA’s prior assessment consultation system, with the aim of shortening the review period for lecanemab. In Europe, Eisai submitted a marketing authorization application (MAA) to the European Medicines Agency (EMA) on January 9, 2023, which was accepted on January 26, 2023. In China, Eisai initiated submission of data for a BLA to the National Medical Products Administration (NMPA) of China in December 2022, and Priority Review was granted on February 27, 2023.
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
* Protofibrils are large Aβ aggregated soluble species of 75-5000 Kd.1
1 Söderberg, L., Johannesson, M., Nygren, P. et al. Lecanemab, Aducanumab, and Gantenerumab – Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics (2022). https://doi.org/10.1007/s13311-022-01308-6. Accessed February 9, 2023
Contacts:
| Eisai |
Biogen Inc. |
MEDIA CONTACT:
Eisai Co., Ltd.
Public Relations Department
TEL: +81 (0)3-3817-5120Eisai Inc. (U.S.)
Libby Holman
+ 1-201-753-1945
Libby_Holman@eisai.com
INVESTOR CONTACT:
Eisai Co., Ltd.
Investor Relations Department
TEL: +81 (0) 3-3817-5122 |
MEDIA CONTACT:
Jack Cox
+ 1-210-544-7920
public.affairs@biogen.com
INVESTOR CONTACT:
Chuck Triano
+ 1-781-464-2442
IR@biogen.com
|
[Notes to editors]
1. About Lecanemab
Lecanemab (Brand Name in the U.S.: LEQEMBI™) is the result of a strategic research alliance between Eisai and BioArctic. Lecanemab is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (protofibril) and insoluble forms of amyloid-beta (Aβ). In the U.S., LEQEMBI was granted accelerated approval by the U.S. Food and Drug Administration (FDA) on January 6, 2023. LEQEMBI is indicated for the treatment of Alzheimer’s disease (AD) in the U.S. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in Aβ plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
Please see full Prescribing Information in the United States.
Eisai has completed lecanemab subcutaneous bioavailability study, and subcutaneous dosing is currently being evaluated in the Clarity AD OLE.
Since July 2020 the Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen. The Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, has been ongoing since January 2022.
2. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
3. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
4. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, our continued commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), is demonstrated by our work on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter, LinkedIn and Facebook.
5. About Biogen
Founded in 1978, Biogen is a leading global biotechnology company that has pioneered multiple breakthrough innovations including a broad portfolio of medicines to treat multiple sclerosis, the first approved treatment for spinal muscular atrophy, and two co-developed treatments to address a defining pathology of Alzheimer’s disease. Biogen is advancing a pipeline of potential novel therapies across neurology, neuropsychiatry, specialized immunology and rare diseases and remains acutely focused on its purpose of serving humanity through science while advancing a healthier, more sustainable and equitable world.
The company routinely posts information that may be important to investors on its website at www.biogen.com. Follow Biogen on social media – Twitter, LinkedIn, Facebook, YouTube.
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements are based on Biogen’s current beliefs and expectations and speak only as of the date of this news release. Biogen does not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
by codm | May 15, 2023 | Newsletter
For Print(PDF)
Listed Company Name: Eisai Co., Ltd.
Representative: Haruo Naito
Representative Corporate Officer and CEO
Headquarters: 4-6-10 Koishikawa,
Bunkyo-ku, Tokyo
Securities Code: 4523; TSE Prime Market
Inquiries: Sayoko Sasaki
Vice President,
Corporate Communications
Phone +81-3-3817-5120
Eisai Co., Ltd. (Headquarters: Tokyo, Representative Corporate Officer and CEO: Haruo Naito, hereinafter “the Company”) announces that a resolution was adopted to revise the Company’s compensation system (the “System”) for directors and corporate officers at a meeting of the Compensation Committee. In addition, at a meeting of the Board of Directors held today, a resolution was adopted for the disposal of treasury stock in accordance with the revision of the stock compensation plan introduced in 2013. Please refer to “Notification Regarding the Disposal of Treasury Stock through Third-Party Allotment in Accordance with the Revision of the Stock Compensation System,” which was announced today, for details regarding the disposal of treasury stock.
Remarks:
1. Revision of the System
The Company has adopted a Nomination Committee, etc. System and the Compensation Committee determines the compensation and related issues with respect to the directors and corporate officers. At the Compensation Committee meeting, the Compensation Committee decided to revise the System as follows:
(1) Directors’ Compensation System
i.Basic concept of the directors’ compensation
The compensation of directors shall be appropriate to enable them to fully perform their duties of management supervision for the common interests of stakeholders and the long-term enhancement of corporate value. Some portion of the compensation of directors shall be paid in the form of shares from the perspective of sharing the same awareness for profits with the shareholders.
ii.Outline of the new compensation system for directors
- The compensation of directors shall be only a fixed amount of base salary, but 90% of the base salary shall be paid in cash and 10% shall be delivered in shares (to be delivered upon retirement).
- Base compensation levels for both external and internal directors will be oriented toward the mid-to-high end of the industry.
- The chairperson of the Board of Directors and the chairpersons of each committee shall be credited with compensation for their services.
(2) Corporate Officers’ Compensation System
I. Basic concept of the corporate officers’ compensation
- Compensation for corporate officers shall be competitive and fully reflect the importance and weight of responsibility of the duties assumed. This will contribute to the realization of the hhc philosophy, attract excellent global human talent, and enhance the morale of the corporate officers in performing their duties.
- Compensation for corporate officers shall be determined with emphasis on the performance and results obtained as a result of implementing business activities for realizing an hhceco company as stipulated in the Articles of Incorporation. This increases the acceptability as compensation for management.
- Compensation for corporate officers shall be based not only on short-term performance based on annual results, but also on strong incentives for corporate officers to enhance the Company’s corporate value, achieve social good, and contribute to social sustainability over the mid- and long-term. In this way, the Company will broadly meet the expectations of stakeholders and contribute to the realization of the Company’s corporate concept.
- Compensation for corporate officers shall be determined by setting appropriate performance targets and incentives that balance “risk, return, and impact” (*1), with objective and appropriate evaluation criteria and a transparent and fair process. This will encourage corporate officers to challenge themselves and provide fair and meaningful compensation, as well as accountability to stakeholders.
(*1) risk (active investment of resources in R&D, etc.), return (company-wide financial performance indicators), and impact (social impact of business activities)
ii. Outline of the new compensation system for corporate officers
- Compensation for corporate officers shall consist of basic compensation (fixed amount) and performance-related compensation (variable), meaning bonuses and stock-based compensation.
- Compensation for corporate officers is set by global job grade (*2) in order to be competitive and reflect the importance and responsibility of the corporate officers’ duties, and the level of compensation is set at the mid-to-high end of the industry.
(*2) A standard that indicates the magnitude of responsibility required for a job and determines compensation.
- Performance-related compensation should be set so that the higher the job grade, the higher the percentage of total compensation.
- Performance-related compensation shall be structured to fully reflect company-wide performance as management compensation, and the ratio of performance-related compensation to total compensation shall aim to be at least 50%. The performance-related compensation will adopt a system in which the results of the corporate officers’ business execution will be evaluated using new indicators, based on the concept of evaluating the results from the perspectives of risk, return, and impact.
- The bonus shall be the sum of the bonus determined based on the degree of achievement of company-wide performance targets (Bonus A) and the bonus determined based on the degree of achievement of individual performance targets (Bonus B), and the ratio of the base amount of calculation between Bonus A and Bonus B shall be 5:5.
The achievement of company-wide performance targets for Bonus A will be determined based on a “risk, return and impact” assessment and will range from 0 to 250%.
The level of achievement of individual performance goals for Bonus B shall be determined based on the evaluation of individual performance goals, including goals related to the realization of the corporate identity stipulated in the Articles of Incorporation, represented by the realization of the social good, and shall be paid in the range of 0 to 150%.
Based on the above, the corporate officers’ bonuses shall range from 0 to 200%.
- The Company will introduce a stock-based compensation system that is linked to mid- to long-term business performance and consists of a portion to be granted during service in office and a portion to be granted upon retirement from office. The ratio of the number of shares to be delivered during service and upon retirement will be 7:3. The portion of shares to be delivered during service will be determined based on ESG EBIT, relative PBR and non-financial company-wide performance targets, and will range from 0 to 150%.
2. Revision of the Stock Compensation System Introduced in Fiscal 2013 (the “Stock Compensation System”) and Additional Contribution to the Officers’ Compensation BIP Trust
In response to the revision of the compensation plan, the Company will revise the Stock Compensation System as follows and make additional contributions to the Officers’ Compensation BIP Trust (the “Trust”) introduced in 2013 after changing the Trust period. The details of the revised Stock Compensation System are as follows.
(1) Outline of the Stock Compensation System
The Stock Compensation System is a stock-based compensation system for directors and corporate officers, under which the Company’s shares are acquired through a trust funded by the amount of compensation for directors and corporate officers contributed by the Company, and through which the Company’s shares and money equivalent to the conversion price of the Company’s shares (the “Company’s Shares and Equivalent”) and an amount equivalent to dividends generated by the Company’s shares are delivered and paid (the “Distribution”). The period under the Stock Compensation System shall be three fiscal years from the fiscal year ending on the last day of March 2024 to the fiscal year ending on the last day of March 2026.
(2) Resolution by the Compensation Committee Relating to the Revision of the Stock Compensation System and Resolution of the Board of Directors relating to the Trust
Since the Company has adopted a Nomination Committee System, the compensation of directors and corporate officers is determined through the Compensation Committee. Therefore, the Compensation Committee adopted a resolution to revise the Stock Compensation System and, thereafter, the Board of Directors also adopted the necessary resolutions to determine the amount of money to be contributed to the Trust, the number of shares to be acquired by the Trust, and other necessary matters.
(3) Subjects of the Stock Compensation System (Beneficiary Requirements)
The subjects of the Stock Compensation System are the directors and corporate officers of the Company, and by completing the prescribed procedures to confirm their status as Beneficiaries, they are eligible to receive the Company’s Shares, etc. each July during the Trust Period on the condition that the following Beneficiary requirements have been met:
<Portion to be delivered upon retirement>
a) During the Trust Period, director or corporate officer must be a party to an engagement contract with the Company;
b) Resignation from any of the Company’s directors or corporate officers;
c) The number of shares to be delivered is determined by the Compensation Committee through the calculation formula set forth in (5) below; and
d) Certain acts of misconduct have not been committed.
<Portion to be delivered during service>
a) During the Trust Period, the corporate officer must be a party to an engagement contract with the Company;
b) The corporate officer must not retire or resign from the Company during the term set forth in the Articles of Incorporation;
c) The number of shares to be distributed is determined by the Compensation Committee through the calculation formula set forth in item (5) below; and
d) Certain acts of misconduct have not been committed.
(4) Post-revision Trust Period
The Trust after the revision will be established by amending and adding to the trust currently established under the Stock Compensation System before the Amendment (the “Existing Trust”) for a trust period of three years from August 1, 2023 (scheduled) to the end of July 2026 (scheduled) (the “Post Revision Trust Period”). With respect to the Existing Trust, although the Company’s shares were acquired under the Stock Compensation Plan before the revision, the Company’s shares remaining in the Existing Trust (“Remaining Shares”) and money (“Remaining Money”) shall be utilized during the post-revision trust period, as the period starting from fiscal 2022 will be reduced from three fiscal years to one fiscal year.
At the end of the Trust Period, the Stock Compensation System can be continued by extending the Trust Period and adding trusts, in accordance with a resolution of the Company’s Compensation Committee and Board of Directors.
In the event of such additional trust, any Remaining Shares and Remaining Money in the trust assets at the end of the trust period prior to the extension, if any, shall be succeeded to the Trust to be extended. This extension of the trust period is not limited to a one-time extension, and the trust period can be extended in the same manner thereafter.
(5) Number of Shares to be Distributed to the Directors and the Corporate Officers (including the number of shares subject to conversion to cash)
The number of shares to be distributed to the directors and the corporate officers (including the number of shares subject to conversion to cash) is to be calculated in accordance with the following formulas for the “portion to be delivered upon retirement” and the “portion to be delivered during service in office”. In the event of a stock split or reverse stock split of the Company’s shares, the number of the Company’s shares to be delivered will be adjusted in accordance with the split ratio, reverse stock split ratio, etc.
Directors
< Portion to be delivered upon retirement >
Number of shares to be delivered upon retirement = Base stock compensation amount by director ÷ Base stock price (*3)
Corporate Officers
< Portion to be delivered upon retirement >
Number of shares to be delivered upon retirement = Base stock-based compensation by job grade × 30% ÷ Base stock price (*3)
< Portion to be delivered during service in office >
Number of shares to be delivered during service = Base stock-based compensation by job grade × 70% ÷ Base stock price (*3) x Performance achievement (*4)
(*3) The base stock price shall be the higher of either the average amount (any fraction less than one yen shall be rounded up) of the closing prices (the “Closing Price”) of the Company’s common stock in regular trading on the Tokyo Stock Exchange on each day (excluding days on which no trading is conducted) of the month containing the Additional Trust Date (May 31, 2023 (scheduled)) or the closing price on the Additional Trust Date (if no closing price on that day, the closing price on the immediately preceding trading day)
(*4) Performance achievement is evaluated and determined by the Compensation Committee based on the achievement of the Company’s ESG EBIT, relative PBR and non-financial and other corporate performance targets for each fiscal year. This determines the share delivery ratio for the number of shares to be delivered at the time of the service, which ranges from 0 to 150%.
(6) The Method and Period of the Distribution of the Company’s Shares and Equivalent to the Directors and Corporate Officers
Directors and corporate officers who fulfill the Beneficiary Requirements shall receive, in principle, 50% of the Company’s shares to be delivered, and for the remaining half shall receive payment of the monetary amount equivalent to the shares converted to cash after they are converted within the Trust. The timing for such Distribution is as follows:
< Portion to be delivered upon retirement >
In principle, upon retirement of directors and corporate officers, they shall receive Distribution of the Company’s shares and Equivalent in a number equivalent to the number of shares delivered upon retirement accumulated during their term of office.
< Portion to be delivered during service in office >
In principle, corporate officers shall receive Distribution of the Company’s shares and Equivalent in July every year during the Trust Period.
It is provided that the directors and corporate officers of the Company will continue to hold the Company’s shares they receive by distribution during their term of office and until one year passes after leaving office.
If a director or a corporate officer dies during the Trust Period, in principle, with respect to the number of all of the Company’s shares for which the Distribution, etc. was determined at such time, the heirs of such director or corporate officer shall receive payment from the Trust of the monetary amount equivalent to the shares converted to cash after they are converted within the Trust.
(7) Amount of Trust Money to be Additionally Entrusted to the Trust and the Number of Shares to be Additionally Acquired by the Trust
The amount of trust money to be additionally entrusted to the Trust and the number of shares to be additionally acquired by the Trust shall be calculated as follows.
Amount of trust money to be additionally entrusted to the Trust:
JPY 1,097,910,000 (*)
*The amount of the total of the above additional trust amount and the Residual Money succeeded from the existing BIP Trust will be allocated to the funds for acquiring shares for the Trust, Trust compensation, and Trust expenses.
Number of shares to be additionally acquired by the Trust:
139,000 Shares (*)
*The number of shares of the total of the above number of shares to be additionally acquired by the Trust and the Residual Shares to be succeeded from the existing BIP Trust will be the number of shares of the Company’s Shares and Equivalent, for which the Distribution to Directors and Corporate Officers is expected during the Trust Period of the Trust.
The amount of money to be additionally entrusted to the Trust will be calculated taking into consideration the amount of compensation of the directors and corporate officers as well as the Trust compensation and Trust expenses.
The number of shares to be additionally acquired shall be set to the level required for distributions in the event of the highest attainment of Company-wide performance objectives, with reference to the present stock price level and the present composition of the directors and corporate officers.
(8) The Method of Acquiring the Company’s Shares through the Trust
After extension, the acquisition of the Company’s shares by the Trust is scheduled to be conducted by way of the disposal of the Company’s treasury stock, in accordance with the number of additionally acquired shares and amount of money for the share acquisitions stipulated in item (7) above.
(9) Exercise of Voting Rights with Regard to the Company’s Shares within the Trust
In order to ensure the neutrality of the Trust’s management, the voting rights of the Company’s shares in the Trust are not to be exercised during the Trust Period.
(10) Handling of the Dividends relating to the Company’s Shares within the Trust
In light of the fact that the Distribution of the Company’s shares and Equivalent for retirement portion will be made at the time of retirement, in order to further raise awareness of contribution to sustainable enhancement of corporate value over the mid- to long-term, dividends paid for the Company’s shares in the Trust will be, as well as used for trust fees and trust expenses of the Trust after the Trust receives them, paid to the beneficiaries together with the Company shares and Equivalent for Distribution in proportion to the number of the Company shares for Distribution (including shares to be converted into cash) from the Trust.
(11) Handling of the Trust at its Expiration
If there are any Residual Shares at the expiration of the Trust Period due to Company-wide performance objectives not being attained during the Trust Period or for other reasons, the Trust will continue to be used as the System or an incentive plan similar thereto by extending the trust agreement and making additional trusts. If the Trust will end due to the expiration of the Trust Period, as a means for sharing and boosting the earnings per share for the shareholders, a gratis transfer of such Residual Shares shall be conducted from the Trust to the Company and the Company is scheduled to cancel the Residual Shares in accordance with a resolution of the Board of Directors. Furthermore, the balance of the dividends related to the Company’s shares within the Trust that exist at the expiration of the Trust Period will be utilized as share acquisition funds if the Trust will continue to be used, but if the Trust will be terminated due to the expiration of the Trust Period, the amount that exceeds the Trust expense reserves are expected to be donated to organizations which have no conflict of interest with the Company or the Company’s directors or corporate officers.
【Details of Trust Agreement】
Outline of the Trust

a) Since the Company has adopted a Nomination Committee System, the Compensation Committee first adopted a resolution to revise the Stock Compensation System, and then the Board of Directors adopted a resolution to dispose of treasury stock in accordance with the Stock Compensation System.
b) In continuing the Stock Compensation System, the Company has established the Basic Policy on Distribution of Shares related compensation of directors and corporate officers.
c) The Company will extend the trust period of the trust which will appoint as beneficiaries those directors and corporate officers who meet the beneficiary requirements (such trust hereinafter the “Trust” and such beneficiaries hereinafter the “Beneficiaries”) in accordance with the resolutions of the Compensation Committee and the Board of Directors as described in item a), and thereafter the Company will entrust additional money.
d) In accordance with the instructions of the Trustee, the Trust will receive the allotment of the Company’s shares (disposal of treasury stock) by the money additionally entrusted in accordance with item c) and the money that remains in the existing Trust. The number of shares the Trust is to acquire shall be set by way of resolution of the Board of Directors in accordance with the resolution of the Compensation Committee as described in item a).
e) Distributions of dividends with respect to the Company’s shares within the Trust will be made similar to other shares.
f) Voting rights for the Company’s shares within the Trust shall not be exercised during the Trust Period.
g) During the Trust Period, the directors and corporate officers who meet the beneficiary requirements will receive a certain proportion of the Company’s shares and the monetary amount equivalent to the shares converted to cash that can be obtained by converting a certain proportion of the Company’s shares, based on Company-wide performance attainment for each fiscal year.
h) If there are any residual shares at the expiration of the trust period due to Company-wide performance objectives not being attained during the trust period, etc., the Trust will continue to be used as the Stock Compensation Plan or an incentive plan similar thereto by extending the trust agreement and making additional trusts, or such shares will be transferred, gratis, from the Trust to the Company, and will be cancelled by way of a resolution of the Board of Directors.
i) Upon the termination of the Trust, the residual assets, after the distribution to the Beneficiaries is completed to the extent that trust expense reserves remain after deducting the stock acquisition funds from the trust money, will belong to the Company. In addition, any amount in excess of the trust expense reserves will be donated to organizations which have no conflict of interest with the Company or the Company’s directors or corporate officers.
(Note) If there are no longer any Company’s shares in the Trust due to the distribution of the Company’s shares or payment of the monetary amounts equivalent to the shares converted for cash to directors and corporate officers that meet the Beneficiaries requirements, the Trust will end before the trust period expires. Furthermore, the Company may entrust additional money to the Trust acquire additional Company shares or trust compensation and to meet Trust expenses.
by codm | May 9, 2023 | Newsletter
For Print(PDF)
Listed Company Name: Eisai Co., Ltd.
Representative: Haruo Naito
Representative Corporate Officer and CEO
Securities Code: 4523
Stock Exchange Listings: Prime Market of the Tokyo Stock Exchange
Inquiries: Sayoko Sasaki
Vice President
Corporate Communications
Phone +81-3-3817-5120
Eisai Co., Ltd. (“the Company”) announced today that based on trends in business results, etc., the Company has revised its consolidated financial forecasts for the fiscal year ending March 31, 2023 (April 1, 2022 to March 31, 2023) previously announced on November 7, 2022, as follows.
1. Revised consolidated financial forecasts for the fiscal year ending March 31, 2023 (April 1, 2022 to March 31, 2023)
(Unit: Millions of yen, unless otherwise noted.)
2. Reason for revision of the consolidated financial forecasts
Based on trends in foreign exchange and product sales, revenue is expected to be ¥744.0 billion, a decrease of ¥16.0 billion from the previous forecast (of which approximately ¥9.5 billion is due to foreign exchange fluctuations).
Due to the decrease in gross profit resulting from the decline in revenue (a decrease of approximately ¥9.5 billion from the previous forecast) and the increase in R&D expenses (an increase of ¥6.5 billion from the previous forecast) resulting from aggressive investments with the good progress of clinical trials for Alzheimer’s disease treatment lecanemab and the review of existing development projects, operating profit is expected to be ¥40.0 billion, a decrease of ¥15.0 billion from the previous forecast.
As a result of the decrease in operating profit and other factors, tax expenses are expected to decrease ¥10.0 billion from the previous forecast, profit for the year is expected to be ¥56.5 billion, a decrease of ¥1.5 billion from the previous forecast, and profit attributable to owners of the parent is expected to be ¥55.0 billion, a decrease of ¥2.0 billion from the previous forecast.
The annual dividend forecast remains unchanged at ¥160 per share, with the year-end dividend of \80 per share (same amount as the previous period) together with the interim dividend (end of the second quarter) of \80 per share, as previously forecast.
* Please note that actual business results may change due to several factors since the above-mentioned forecasts were made based on information available as of May 9, 2023.






