
by codm | Jan 7, 2023 | Newsletter
MAXIMIZING VALUE FOR ALL STAKEHOLDERS WHILE GIVING BACK VALUE TO SOCIETY
For Print(PDF)(364KB)
Based on hhc (human health care) concept, our corporate philosophy, Eisai is committed to improve patients health outcomes and quality of life, simplify care delivery, increase health system efficiency and spur future investments in Alzheimer’s disease (AD). We consider a holistic approach in assessing value and making decisions that may affect patient access, so that our LEQEMBI pricing approach can maximize value for all stakeholders (patients, families, caregivers, healthcare providers, payers, employees and shareholders). This approach includes clinical outcome assessments of our medicines and the benefits we deliver to patients, their families and caregivers as the “clinical value,” as well as the projected “social value” that help improve patients’ and caregivers’ quality of life and productivity. Moreover, we assess the simulated impact of our medicines on reducing demand for health services and global burden of disease as potential “economic value” while enhancing further innovations in AD.
Social Impact of AD in the U.S.
According to the Alzheimer’s Association’s “2022 Alzheimer’s Disease Facts and Figures”1, an estimated 6.5 million Americans aged 65 and older are living with dementia due to AD (i.e., mild, moderate and severe dementia stages of AD). AD was officially listed as the sixth-leading cause of death in the U.S. in 2019 and the seventh-leading cause of death in 2020 and 2021. It is a chronic, progressive, disabling and fatal disease. According to another report from the Alzheimer’s Association, “Changing the Trajectory of Alzheimer’s Disease: How a Treatment by 2025 Saves Lives and Dollars”2, the total costs of care in the U.S. from all payers (i.e., Medicare, Medicaid, out-of-pocket and other payers), would increase from $267 Billion in 2020 to $451 Billion in 2030 if no treatment exists to delay the disease.
A recent 2022 study, “Projecting the Long-term Societal Values of Disease-Modifying Treatment for Alzheimer’s Disease in the United States”3, quoted as many as 10 to 14 million Americans living with MCI (Mild Cognitive Impairment) of any etiology, the very mild symptomatic stage before onset of dementia stages, in which 55% have AD as the underlying pathology. In this study, the lifetime value of a disease-modifying treatment in Early AD (MCI due to AD and mild dementia stage of AD) from a U.S. societal perspective assuming a treatment effect of 30% relative decline in progression rates from MCI to mild dementia and from mild to moderate dementia was estimated at $134,418 per person.
Value of LEQEMBI Adoption to U.S. Society
LEQEMBI is indicated for the treatment of AD, and treatment with LEQEMBI should be initiated in patients with MCI or mild dementia stage of AD after confirmation of amyloid beta pathology (Early AD). In the U.S., we estimate that the diagnosed eligible Early AD population will reach approximately 100,000 individuals by year 3 representing a measured initial attainment in the real world and will increase gradually over the mid-to-long term given the time required to advance new screening and diagnostic technologies such as blood-based biomarkers to confirm amyloid beta pathology.
Published findings in a peer-reviewed journal4 from the confirmatory Phase 3 Clarity AD study in patients with Early AD demonstrate that LEQEMBI treatment resulted in less decline on measures of cognition and function than placebo at 18 months (27% slowing over 18 months measured by CDR-SB*) and was associated with adverse events that were within expectation. Clarity AD results were consistent with that from the Phase 2 trial (Study 201)5 that was the basis for the FDA’s accelerated approval and will be submitted to the FDA very shortly for review for traditional approval.
LEQEMBI’s clinical data show that it could help patients maintain cognition, preserve activities of daily living and maintain functional ability for longer, and therefore, LEQEMBI’s clinical efficacy could potentially translate into impactful outcomes for these patients and their families. A simulation study using an established, validated disease model called Alzheimer’s disease Archimedes condition event (AD ACE),6,7,8 which is an individual patient-level model with a focus on predicting the trajectory of cognitive decline and simulating the effects of early interventions in AD, helped assess the potential lifetime value and economic impact of LEQEMBI in patients with Early AD based on clinical trial populations and findings. The results9,10 of this simulation study based on Study 201 of LEQEMBI were published in a peer-reviewed journal, and the model has been recently updated with data from the Clarity AD trial showing consistent outputs.
Per Patient Societal Value of LEQEMBI at $37,600 per Year in the U.S. by AD ACE Model
In the updated AD ACE simulation study using Clarity AD data, LEQEMBI treatment was projected to delay disease progression, resulting in an increase in patient’s expected time in Early AD while reducing the time in more advanced severe states. Slowing of clinical decline in patients treated with LEQEMBI is estimated to delay disease progression by nearly 3 years on average (delay progression mainly from MCI to mild AD and from mild to moderate AD) compared to standard-of-care (SOC). LEQEMBI’s impact on the disease trajectory is then modeled into an annual per-patient value to the U.S. society based on four components according to the following equation: a) quality-adjusted life-years (QALY) gains compared to SOC; b) willingness-to-pay (WTP) threshold; c) cost offsets compared to SOC; and d) time on treatment, all in present value term. Note that the costs, benefits and time on treatment need to be adjusted for the values between present time and future times using discounting in a health economic evaluation.
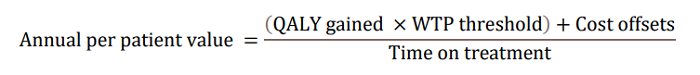
With regard to a) QALY gains, since health is a function of length of life and quality of life, this measure combines both attributes into one single index, with one QALY gain representing one additional year of a person’s life at perfect health. LEQEMBI treatment was predicted to offer an additional 0.64 QALYs compared to SOC for an Early AD patient over lifetime by improving outcomes for both the patient and caregiver. With regard to b) WTP threshold per QALY gained, conventionally, the WTP threshold is based on 1 to 3 times country’s per capita gross domestic product (GDP). In the U.S., as a result, a WTP threshold of $50,000 to $150,000 is referenced as the cost-effectiveness threshold11, but a higher WTP threshold is often considered to account for the gravity of conditions with greater burden, and interventions that exhibit wider societal benefits, such as AD with substantial impacts on caregivers. A modified societal perspective at $200,000 WTP threshold per QALY gained is used in the U.S. when societal cost of the disease is large such as AD. With regard to c) cost offsets, avoidance of both direct medical/non-medical costs for AD management, as well as indirect costs of caregivers including family members totaling $7,415 over the lifetime for each Early AD patient, was predicted with LEQEMBI treatment compared to SOC. Direct costs contain cost of medications, medical visits, hospitalizations, living accommodations and community services for the patients. Indirect costs of caregivers, including family members, consider the monetary value for hours spent on caregiving activities. With regard to: d) time on treatment, LEQEMBI was modeled to be stopped upon transition to moderate AD dementia or worse. The average treatment duration in this Early AD population was estimated to be approximately 3.6 years with consideration of discounting to present value.
Taking all four components together in present value term, for a) 0.64 QALYs gained b) at WTP threshold of $200,000 per QALY gained plus c) cost offsets of $7,415 over d) 3.6 years on treatment, the yearly per-patient value of LEQEMBI from a societal perspective was quantified at approximately $37,600.
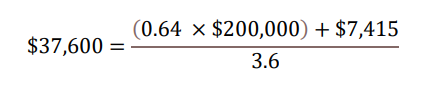
This yearly estimate of $37,600 equals to approximately $135,000 (0.64 × $200,000 + $7,415) in lifetime value per patient to the U.S. society. Over 10 years cumulatively, the gradual adoption of LEQEMBI treatment among Early AD patients could potentially generate positive social impact of several tens of billion dollars to the U.S. society, in the form of “clinical value” that help patients delay their disease progression, projected “social value” that help improve patients’ and caregivers’ quality of life and productivity, and simulated “economic value” that help reduce demand for health services.
LEQEMBI U.S. Launch Pricing at $26,500 per Year
While we estimate the per-patient-per-year value of LEQEMBI treatment to the U.S. society to be $37,600, Eisai decided to price LEQEMBI below quantified societal value at the wholesale acquisition cost (WAC) of $26,500 per year (estimated annual price based on 10mg/kg IV biweekly for average U.S. patient weight of 75kg based on Study 201 and Clarity AD) aiming to promote broader patient access, reduce overall financial burden, and support health system sustainability. As such, the WAC for the 200mg vial is $254.81 and the WAC for the 500mg vial is $637.02. Actual annualized pricing may vary by patient. In addition, Eisai continues to pursue a less frequent maintenance dosing regimen for LEQEMBI, such as monthly instead of current biweekly regimen, upon significant amyloid beta clearance to prevent re-accumulation of amyloid beta biomarkers while maintaining clinical efficacy. This could further lower the yearly cost of LEQEMBI during the maintenance dosing phase, for example, from $26,500 to potentially about half of this figure given less amount of drugs.
Patient Affordability
Eisai believes patient affordability must be a key consideration to promote patient access and intended use and benefits of LEQEMBI. Among the eligible Early AD patient population in the U.S., once the patient’s insurer covers LEQEMBI, we estimate that approximately 91% of individuals will be covered by Medicare with Medigap (supplemental insurance), Medicare Advantage (Medicare-approved plans from private companies with potential supplemental coverage), Medicaid, and Commercial (private insurance12). For these patients, their out-of-pocket costs for LEQEMBI treatment could range from $0 to a few dollars per day. Remaining 9% of the individuals will fall into the category of Medicare without supplemental insurance, and hence will be responsible for 20% of the LEQEMBI cost as co-insurance under Medicare Part B. For these patients, their estimated out-of-pocket costs for LEQEMBI at the price of $26,500 per year will translate into about $14.50 per day13. Across the entire eligible Early AD patient population, we estimate the weighted average out-of-pocket costs for LEQEMBI to be about $2 per day12.
Commitment to Patient Access
Eisai is committed to ensuring that certain financially disadvantaged patients have access to LEQEMBI. Firstly, Eisai is establishing a Patient Assistance Program, which will provide LEQEMBI at no cost, for eligible uninsured and underinsured patients, including Medicare beneficiaries, who meet financial need and other program criteria. Secondly, Eisai will offer patient support for improving access through LEQEMBI Patient Navigators, who will provide information about accessing LEQEMBI, help patients and their families understand their insurance coverage and options, and identify financial support programs for eligible patients.
Health System Sustainability
We believe our pricing approach for LEQEMBI would also help improve health system sustainability, which is projected based on appropriate use of LEQEMBI in eligible patients with Early AD to improve patient’s health outcomes and quality of life and reduce demand for health services and global burden of disease through changing disease trajectory. Furthermore, we believe our pricing approach for LEQEMBI, coupled with the size of the targeted patient population, will be sustainable under historical growth and spending assumptions for Medicare Part B.
Giving Back More Than Half of LEQEMBI Value to U.S. Society
The price of LEQEMBI at a yearly cost of $26,500 is $11,100 below the projected societal value of $37,600, and a less frequent maintenance dosing regimen will further lower the yearly cost well below the projected societal value. Taking these savings as well as discounts and rebates within the U.S. healthcare system into consideration, over 10 years cumulatively, the gradual adoption of LEQEMBI treatment at this pricing approach could give back about 60% of the potential positive social impact of several tens of billion dollars to the U.S. society14. These resources could help realize new innovations that enhance the health and quality of life of individuals at risk of developing or living with AD as well as their families and caregivers. On the other hand, about 40% of the potential positive social impact of several tens of billion dollars will be accrued by employees and shareholders in the form of product sales, from which we are committed to re-invest in future research and development to create new AD therapies and new innovations such as establishing ecosystems toward inclusive AD communities14. We deeply believe that our pricing approach to maximize value for all stakeholders will help Eisai achieve social good in the form of relieving anxiety over health and reducing health disparity according to our corporate philosophy.
*Clinical Dementia Rating Sum of Boxes
(End of Document)
Notes to Editors
INDICATION, DOSAGE AND ADMINISTRATION, AND IMPORTANT SAFETY INFORMATION IN THE U.S.
INDICATION
LEQEMBI is indicated for the treatment of Alzheimer’s disease. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Amyloid Related Imaging Abnormalities
- LEQEMBI can cause amyloid related imaging abnormalities-edema (ARIA-E) and -hemosiderin deposition (ARIA-H). ARIA-E can be observed on MRI as brain edema or sulcal effusions, and ARIA-H as microhemorrhage and superficial siderosis. ARIA is usually asymptomatic, although serious and life-threatening events, including seizure and status epilepticus, rarely can occur. Reported symptoms associated with ARIA may include headache, confusion, visual changes, dizziness, nausea, and gait difficulty. Focal neurologic deficits may also occur. Symptoms associated with ARIA usually resolve over time.
ARIA Monitoring and Dose Management Guidelines
- Obtain recent (within one year) brain magnetic resonance imaging (MRI) prior to initiating treatment with LEQEMBI. Obtain an MRI prior to the 5th, 7th, and 14th infusions.
- Recommendations for dosing in patients with ARIA-E and ARIA-H depend on clinical symptoms and radiographic severity. Depending on ARIA severity, use clinical judgment in considering whether to continue dosing, temporarily discontinue treatment, or permanently discontinue LEQEMBI.
- Enhanced clinical vigilance for ARIA is recommended during the first 14 weeks of treatment with LEQEMBI. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI if indicated. If ARIA is observed on MRI, careful clinical evaluation should be performed prior to continuing treatment.
- There is no experience in patients who continued dosing through symptomatic ARIA-E or through asymptomatic, but radiographically severe, ARIA-E. There is limited experience in patients who continued dosing through asymptomatic but radiographically mild to moderate ARIA-E. There are limited data in dosing patients who experienced recurrent ARIA-E.
Incidence of ARIA
- In Study 1 (Study 201), symptomatic ARIA occurred in 3% (5/161) of LEQEMBI-treated patients. Clinical symptoms associated with ARIA resolved in 80% of patients during the period of observation.
- Including asymptomatic cases, ARIA was observed in LEQEMBI: 12% (20/161); placebo: 5% (13/245). ARIA-E was observed in LEQEMBI: 10% (16/161); placebo: 1% (2/245). ARIA-H was observed in LEQEMBI: 6% (10/161); placebo: 5% (12/245). There was no increase in isolated ARIA-H for LEQEMBI compared to placebo.
- Intracerebral hemorrhage >1 cm in diameter was reported after one treatment in LEQEMBI: 1 patient; placebo: zero patients. Events of intracerebral hemorrhage, including fatal events, in patients taking LEQEMBI have also been reported in other studies.
Apolipoprotein E ε4 (ApoE ε4) Carrier Status and Risk of ARIA
- In Study 1, 6% (10/161) of patients in the LEQEMBI group were ApoE ε4 homozygotes, 24% (39/161) were heterozygotes, and 70% (112/161) were noncarriers.
- The incidence of ARIA was higher in ApoE ε4 homozygotes than in heterozygotes and noncarriers among patients treated with LEQEMBI. Of the 5 LEQEMBI-treated patients who had symptomatic ARIA, 4 were ApoE ε4 homozygotes, 2 of whom experienced severe symptoms. An increased incidence of symptomatic and overall ARIA in ApoE ε4 homozygotes compared to heterozygotes and noncarriers in LEQEMBI-treated patients has been reported in other studies.
- The recommendations on management of ARIA do not differ between ApoE ε4 carriers and noncarriers.
- Consider testing for ApoE ε4 status to inform the risk of developing ARIA when deciding to initiate treatment with LEQEMBI.
Radiographic Findings
- The majority of ARIA-E radiographic events occurred early in treatment (within the first 7 doses), although ARIA can occur at any time and patients can have more than 1 episode. The maximum radiographic severity of ARIA-E in patients treated with LEQEMBI was mild in 4% (7/161) of patients, moderate in 4% (7/161) of patients, and severe in 1% (2/161) of patients. Resolution on MRI occurred in 62% of ARIA-E patients by 12 weeks, 81% by 21 weeks, and 94% overall after detection. The maximum radiographic severity of ARIA-H microhemorrhage in patients treated with LEQEMBI was mild in 4% (7/161) of patients and severe in 1% (2/161) of patients; 1 of the 10 patients with ARIA-H had mild superficial siderosis.
Concomitant Antithrombotic Medication and Other Risk Factors for Intracerebral Hemorrhage
- Patients were excluded from enrollment in Study 1 for baseline use of anticoagulant medications. Antiplatelet medications such as aspirin and clopidogrel were allowed. If anticoagulant medication was used because of intercurrent medical events that required treatment for ≤4 weeks, treatment with LEQEMBI was to be temporarily suspended.
- Most exposures to antithrombotic medications were to aspirin; few patients were exposed to other antiplatelet drugs or anticoagulants, limiting any meaningful conclusions about the risk of ARIA or intracerebral hemorrhage in patients taking other antiplatelet drugs or anticoagulants. Because intracerebral hemorrhages >1 cm in diameter have been observed in patients taking LEQEMBI, additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with LEQEMBI.
- Patients were excluded from enrollment in Study 1 for the following risk factors for intracerebral hemorrhage: prior cerebral hemorrhage >1 cm in greatest diameter, more than 4 microhemorrhages, superficial siderosis, evidence of vasogenic edema, evidence of cerebral contusion, aneurysm, vascular malformation, infective lesions, multiple lacunar infarcts or stroke involving a major vascular territory, and severe small vessel or white matter disease. Caution should be exercised when considering the use of LEQEMBI in patients with these risk factors.
Infusion-Related Reactions
- Infusion-related reactions were observed in LEQEMBI: 20% (32/161); placebo: 3% (8/245), and the majority of cases in LEQEMBI-treated patients (88%, 28/32) occurred with the first infusion. All infusion-related reactions were mild (56%) or moderate (44%) in severity. Infusion-related reactions resulted in discontinuations in 2% (4/161) of patients treated with LEQEMBI. Symptoms of infusion-related reactions included fever and flu-like symptoms (chills, generalized aches, feeling shaky, and joint pain), nausea, vomiting, hypotension, hypertension, and oxygen desaturation.
- After the first infusion, 38% of LEQEMBI-treated patients had transient decreased lymphocyte counts to <0.9 x109/L compared to 2% on placebo, and 22% of LEQEMBI-treated patients had transient increased neutrophil counts to >7.9 x109/L compared to 1% on placebo.
- In the event of an infusion-related reaction, the infusion rate may be reduced, or the infusion may be discontinued, and appropriate therapy initiated as clinically indicated. Prophylactic treatment with antihistamines, acetaminophen, nonsteroidal anti-inflammatory drugs, or corticosteroids prior to future infusions may be considered.
ADVERSE REACTIONS
- In Study 1, 15% of LEQEMBI-treated patients, compared to 6% of placebo-treated patients, stopped study treatment because of an adverse reaction. The most common adverse reaction leading to discontinuation of LEQEMBI was infusion-related reactions that led to discontinuation in 2% (4/161) of patients treated with LEQEMBI compared to 1% (2/245) of patients on placebo.
- The most common adverse reactions reported in ≥5% of patients treated with LEQEMBI (N=161) and ≥2% higher than placebo (N=245) in Study 1 were infusion-related reactions (LEQEMBI: 20%; placebo: 3%), headache (LEQEMBI: 14%; placebo: 10%), ARIA-E (LEQEMBI: 10%; placebo: 1%), cough (LEQEMBI: 9%; placebo: 5%), and diarrhea (LEQEMBI: 8%; placebo: 5%).
Please see full Prescribing Information
- Alzheimer’s Association, 2022 Alzheimer’s Disease Facts and Figures
https://www.alz.org/media/Documents/alzheimers-facts-and-figures.pdf
- Alzheimer’s Association, Changing the Trajectory of Alzheimer’s Disease
https://www.alz.org/media/documents/changing-the-trajectory-r.pdf
- Prados M, et al. Projecting the long‐term societal value of a disease‐modifying treatment for Alzheimer’s disease in the United States. Alzheimers Dement. 2022; 18(1):142-151. doi: 10.1002/alz.12578. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9303743/
- van Dyck. C, et al. Lecanemab in Early Alzheimer’s Disease. The New England Journal of Medicine. DOI: 10.1056/NEJMoa2212948.
https://www.nejm.org/doi/full/10.1056/NEJMoa2212948
- Swanson CJ, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody
https://alzres.biomedcentral.com/articles/10.1186/s13195-021-00813-8
- Kansal AR, Tafazzoli A, Ishak KJ, Krotneva S. Alzheimer’s disease Archimedes condition-event simulator: Development and validation. Alzheimers Dement (NY). 2018;4:76-88. Published 2018 Feb 16. doi:10.1016/j.trci.2018.01.001
- Tafazzoli A, and Kansal A. Disease simulation in drug development, External validation confirms benefit in decision making. The Evidence Forum. 2018.
https://www.evidera.com/wp-content/uploads/2018/10/07-Disease-Simulation-in-Drug-Development_Fall2018.pdf
- Tafazzoli A, Weng J, Sutton K, et al. Validating simulated cognition trajectories based on ADNI against 436 trajectories from the National Alzheimer’s Coordinating Center (NACC) dataset. 11th edition of Clinical Trials on 437 Alzheimer’s Disease (CTAD); Barcelona, Spain: 2018.
- Tahami Monfared AA, et al. Long-term health outcomes of lecanemab in patients with early Alzheimer’s disease using simulation modeling. Neurol Ther. 2022;11:863–880.
https://link.springer.com/article/10.1007/s40120-022-00350-y
- Tahami Monfared AA, et al. The Potential Economic Value of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling, Neurol Ther. 2022;11: 1285–1307.
https://link.springer.com/article/10.1007/s40120-022-00373-5
- ICER Value Framework 2020-2023. 2022.
https://icer.org/wp-content/uploads/2020/11/ICER_2020_2023_VAF_02032022.pdf
- IQVIA. Longitudinal Access and Adjudication DATA (LAAD) 2019-2021
- 20% of the WAC price ($26,500) divided by 365 (days)
- Internal Eisai calculations, December 2022
Safe Harbor Statement
Materials and information provided in this announcement may contain so-called “forward-looking statements.” These statements are based on current expectations, forecasts and assumptions that are subject to risks and uncertainties that could cause actual outcomes and results to differ materially from these statements.
Risks and uncertainties include general industry and market conditions, and general domestic and international economic conditions such as interest rate and currency exchange fluctuations. Risks and uncertainties particularly apply with respect to product-related forward-looking statements. Product risks and uncertainties include, but are not limited to, technological advances and patents attained by competitors; challenges inherent in new product development, including completion of clinical trials; claims and concerns about product safety and efficacy; regulatory agency examination periods and obtaining regulatory approvals; domestic and foreign healthcare reforms; trends toward managed care and healthcare cost containment; and governmental laws and regulations affecting domestic and foreign operations.
The Company cannot guarantee the actual outcomes and results for any forward-looking statements.
The Company disclaims any intention or obligation to update or revise any forward-looking statements whether as a result of new information, future events or otherwise.
For Print(PDF)(364KB)
by codm | Jan 7, 2023 | Newsletter
Accelerated Approval is based on Phase 2 data showing a reduction in amyloid-beta plaques in early AD patients treated with LEQEMBI™
Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials
For Print(PDF)(243KB)
TOKYO and CAMBRIDGE, Mass., January 7, 2023 – Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, “Biogen”) announced today that under the Accelerated Approval Pathway the U.S. Food and Drug Administration (FDA) has approved lecanemab-irmb (Brand Name in the U.S.: LEQEMBI™) 100 mg/mL injection for intravenous use, a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (“protofibril”)* and insoluble forms of amyloid beta (Aβ) for the treatment of Alzheimer’s disease (AD). The approval is based on Phase 2 data that demonstrated that LEQEMBI reduced the accumulation of Aβ plaque in the brain, a defining feature of AD. Using the recently published data from the large global confirmatory Phase 3 clinical trial, Clarity AD, Eisai will work quickly to file a Supplemental Biologics License Application (sBLA) to the FDA for approval under the traditional pathway.
INDICATION
LEQEMBI is indicated for the treatment of Alzheimer’s disease. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
DOSAGE AND ADMINISTRATION (Patient Selection, Dosing Instructions, Monitoring and Dosing Interruption for ARIA)
The recommended dosage of LEQEMBI is 10 mg/kg administered intravenously once every two weeks to eligible patients with confirmed presence of Aβ pathology prior to initiating treatment. Enhanced clinical vigilance for amyloid-related imaging abnormalities (ARIA) is recommended during the first 14 weeks of treatment with LEQEMBI. Baseline, recent (within one year) brain MRI prior to initiating treatment with LEQEMBI and periodic monitoring with MRI prior to the 5th, 7th, and 14th infusions should be obtained.
ADVERSE REACTIONS
The safety of LEQEMBI has been evaluated in 763 patients who received at least one dose of LEQEMBI in Study 201. The most common adverse reactions reported in at least 5% of patients treated with LEQEMBI 10 mg/kg biweekly (N=161) and at least 2% higher incidence than patients on placebo (N=245) were infusion-related reactions (LEQEMBI 20%; placebo 3%), headache (LEQEMBI 14%; placebo 10%), ARIA-E (LEQEMBI 10%; placebo 1%), cough (LEQEMBI, 9%; placebo, 5%) and diarrhea (LEQEMBI, 8%; placebo, 5%). The most common adverse reaction leading to discontinuation of LEQEMBI was infusion-related reactions that led to discontinuation in 2% (4/161) of patients treated with LEQEMBI compared to 1% (2/245) of patients on placebo.
CONCOMITANT ANTITHROMBOTIC MEDICATION AND OTHER RISK FACTORS FOR INTRACEREBRAL HEMORRHAGE
Patients were excluded from enrollment in Study 201 for baseline use of anticoagulant medications. Antiplatelet medications such as aspirin and clopidogrel were allowed. Patients who received LEQEMBI and an antithrombotic medication (aspirin, other antiplatelets, or anticoagulants) did not have an increased risk of ARIA-H compared to patients who received placebo and an antithrombotic medication. The majority of exposures to antithrombotic medications were to aspirin; few patients were exposed to other antiplatelet drugs or anticoagulants, limiting any meaningful conclusions about the risk of ARIA or intracerebral hemorrhage in patients taking other antiplatelet drugs or anticoagulants. Because intracerebral hemorrhages greater than 1 cm in diameter have been observed in patients taking LEQEMBI, additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with LEQEMBI. Additionally, patients were excluded from enrollment in Study 201 for the following risk factors for intracerebral hemorrhage: prior cerebral hemorrhage greater than 1 cm in greatest diameter, more than 4 microhemorrhages, superficial siderosis, evidence of vasogenic edema, evidence of cerebral contusion, aneurysm, vascular malformation, infective lesions, multiple lacunar infarcts or stroke involving a major vascular territory, and severe small vessel or white matter disease. Caution should be exercised when considering the use of LEQEMBI in patients with these risk factors.
“The FDA’s approval of LEQEMBI under the Accelerated Approval pathway is an important milestone in Eisai’s four decades of research in Alzheimer’s disease and reflects our continued commitment to alleviating the burden of Alzheimer’s disease for patients and their families. Eisai has made great efforts to understand the reality of the challenges and concerns facing patients and their families who are living in the various stages of Alzheimer’s disease, and we are incredibly pleased to offer LEQEMBI as a new treatment option to help with the tremendous unmet needs of this community,” said Haruo Naito, Chief Executive Officer at Eisai Co., Ltd. “The challenges of Alzheimer’s disease reach beyond medical implications for patients and considerations for their families, but also impact society as a whole through reduced productivity, elevated social costs and anxiety. Upon receiving this Accelerated Approval, we will focus on providing important information on proper usage of LEQEMBI to healthcare professionals. Eisai will also engage with various payers to provide access to LEQEMBI, offer a patient support program, and will do its utmost to complete submission for traditional approval as soon as possible to serve more people living with early Alzheimer’s disease.”
“The approval of LEQEMBI provides new hope to patients with Alzheimer’s disease. Patients at an early stage of the disease and their caregivers can now consider a new treatment option with their doctors. Our focus now is on the path forward, working alongside Eisai with the goal of making LEQEMBI available to patients who may benefit from this treatment as soon as possible,” said Christopher A. Viehbacher, President and Chief Executive Officer of Biogen. “This approval is also a recognition of the many scientists and doctors who have, over many years, patiently and persistently worked to find a treatment for this highly complex disease. Eisai and Biogen have collaborated for nearly a decade to advance research to improve the lives of those suffering from Alzheimer’s, and we know that this commitment must and will continue in the fight against Alzheimer’s disease.”
LEQEMBI’s ACCESS AND INITIATIVES TO SUPPORT PEOPLE LIVING WITH AD
The Eisai Patient Support Program offers several support programs to help patients and care partners. Dedicated Patient Navigators will work directly with patients and families to navigate treatment and coverage for eligible and appropriate patients and to help with what to expect regarding insurance coverage, co-pay and patient access programs. To learn more visit LEQEMBI.com(New Window), call 1-833-4-LEQEMBI (1-833-453-7362), Monday-Friday, 8 a.m. to 8 p.m. Eastern Time or fax to 1-833-770-7017.
In addition, to support access to LEQEMBI for certain financially disadvantaged patients, Eisai’s Patient Assistance Program (PAP) will provide LEQEMBI at no cost, for eligible uninsured and underinsured patients, including Medicare beneficiaries, who meet financial need and other program criteria.
Eisai looks forward to continuing to engage constructively with various payors, including the Centers for Medicare and Medicaid (CMS), TRICARE, the U.S. Veteran’s Health Administration and private health insurance companies to ensure appropriate beneficiaries have access to this new therapy. Currently, Medicare patients do not have access to LEQEMBI. Medicaid sole beneficiaries who are diagnosed by a healthcare professional with mild cognitive impairment or mild dementia stage of disease, and with confirmed presence of amyloid plaque in the brain will have access to LEQEMBI under the Medicaid program post accelerated approval, depending on individual state processes.
Eisai is developing a multi-faceted educational initiative to further advance the understanding in the AD healthcare community of the real-world management and monitoring of ARIA. This initiative, Understanding ARIA™, will provide resources and programs that will include peer-to-peer education, individual and group educational sessions and subject-matter-expert evaluation of historical case studies. Understanding ARIA will include engagements with leading experts in medical imaging as well as major professional societies. Initial resources will be available by January 2023.
LEQEMBI will be available during or before the week of January 23, 2023. Eisai announced the U.S. pricing and rationale for LEQEMBI today.
Eisai serves as the lead of LEQEMBI development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
*Protofibrils are large Aβ aggregated soluble species of 75-500 Kd. 1, 2
INDICATION, DOSAGE AND ADMINISTRATION, AND IMPORTANT SAFETY INFORMATION IN THE U.S.
INDICATION
LEQEMBI is indicated for the treatment of Alzheimer’s disease. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Amyloid Related Imaging Abnormalities
- LEQEMBI can cause amyloid related imaging abnormalities-edema (ARIA-E) and -hemosiderin deposition (ARIA-H). ARIA-E can be observed on MRI as brain edema or sulcal effusions, and ARIA-H as microhemorrhage and superficial siderosis. ARIA is usually asymptomatic, although serious and life-threatening events, including seizure and status epilepticus, rarely can occur. Reported symptoms associated with ARIA may include headache, confusion, visual changes, dizziness, nausea, and gait difficulty. Focal neurologic deficits may also occur. Symptoms associated with ARIA usually resolve over time.
ARIA Monitoring and Dose Management Guidelines
- Obtain recent (within one year) brain magnetic resonance imaging (MRI) prior to initiating treatment with LEQEMBI. Obtain an MRI prior to the 5th, 7th, and 14th infusions.
- Recommendations for dosing in patients with ARIA-E and ARIA-H depend on clinical symptoms and radiographic severity. Depending on ARIA severity, use clinical judgment in considering whether to continue dosing, temporarily discontinue treatment, or permanently discontinue LEQEMBI.
- Enhanced clinical vigilance for ARIA is recommended during the first 14 weeks of treatment with LEQEMBI. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI if indicated. If ARIA is observed on MRI, careful clinical evaluation should be performed prior to continuing treatment.
- There is no experience in patients who continued dosing through symptomatic ARIA-E or through asymptomatic, but radiographically severe, ARIA-E. There is limited experience in patients who continued dosing through asymptomatic but radiographically mild to moderate ARIA-E. There are limited data in dosing patients who experienced recurrent ARIA-E.
Incidence of ARIA
- In Study 1 (Study 201), symptomatic ARIA occurred in 3% (5/161) of LEQEMBI-treated patients. Clinical symptoms associated with ARIA resolved in 80% of patients during the period of observation.
- Including asymptomatic cases, ARIA was observed in LEQEMBI: 12% (20/161); placebo: 5% (13/245). ARIA-E was observed in LEQEMBI: 10% (16/161); placebo: 1% (2/245). ARIA-H was observed in LEQEMBI: 6% (10/161); placebo: 5% (12/245). There was no increase in isolated ARIA-H for LEQEMBI compared to placebo.
- Intracerebral hemorrhage >1 cm in diameter was reported after one treatment in LEQEMBI: 1 patient; placebo: zero patients. Events of intracerebral hemorrhage, including fatal events, in patients taking LEQEMBI have also been reported in other studies.
Apolipoprotein E ε4 (ApoE ε4) Carrier Status and Risk of ARIA
- In Study 1, 6% (10/161) of patients in the LEQEMBI group were ApoE ε4 homozygotes, 24% (39/161) were heterozygotes, and 70% (112/161) were noncarriers.
- The incidence of ARIA was higher in ApoE ε4 homozygotes than in heterozygotes and noncarriers among patients treated with LEQEMBI. Of the 5 LEQEMBI-treated patients who had symptomatic ARIA, 4 were ApoE ε4 homozygotes, 2 of whom experienced severe symptoms. An increased incidence of symptomatic and overall ARIA in ApoE ε4 homozygotes compared to heterozygotes and noncarriers in LEQEMBI-treated patients has been reported in other studies.
- The recommendations on management of ARIA do not differ between ApoE ε4 carriers and noncarriers.
- Consider testing for ApoE ε4 status to inform the risk of developing ARIA when deciding to initiate treatment with LEQEMBI.
Radiographic Findings
- The majority of ARIA-E radiographic events occurred early in treatment (within the first 7 doses), although ARIA can occur at any time and patients can have more than 1 episode. The maximum radiographic severity of ARIA-E in patients treated with LEQEMBI was mild in 4% (7/161) of patients, moderate in 4% (7/161) of patients, and severe in 1% (2/161) of patients. Resolution on MRI occurred in 62% of ARIA-E patients by 12 weeks, 81% by 21 weeks, and 94% overall after detection. The maximum radiographic severity of ARIA-H microhemorrhage in patients treated with LEQEMBI was mild in 4% (7/161) of patients and severe in 1% (2/161) of patients; 1 of the 10 patients with ARIA-H had mild superficial siderosis.
Concomitant Antithrombotic Medication and Other Risk Factors for Intracerebral Hemorrhage
- Patients were excluded from enrollment in Study 1 for baseline use of anticoagulant medications. Antiplatelet medications such as aspirin and clopidogrel were allowed. If anticoagulant medication was used because of intercurrent medical events that required treatment for ≤4 weeks, treatment with LEQEMBI was to be temporarily suspended.
- Most exposures to antithrombotic medications were to aspirin; few patients were exposed to other antiplatelet drugs or anticoagulants, limiting any meaningful conclusions about the risk of ARIA or intracerebral hemorrhage in patients taking other antiplatelet drugs or anticoagulants. Because intracerebral hemorrhages >1 cm in diameter have been observed in patients taking LEQEMBI, additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with LEQEMBI.
- Patients were excluded from enrollment in Study 1 for the following risk factors for intracerebral hemorrhage: prior cerebral hemorrhage >1 cm in greatest diameter, more than 4 microhemorrhages, superficial siderosis, evidence of vasogenic edema, evidence of cerebral contusion, aneurysm, vascular malformation, infective lesions, multiple lacunar infarcts or stroke involving a major vascular territory, and severe small vessel or white matter disease. Caution should be exercised when considering the use of LEQEMBI in patients with these risk factors.
Infusion-Related Reactions
- Infusion-related reactions were observed in LEQEMBI: 20% (32/161); placebo: 3% (8/245), and the majority of cases in LEQEMBI-treated patients (88%, 28/32) occurred with the first infusion. All infusion-related reactions were mild (56%) or moderate (44%) in severity. Infusion-related reactions resulted in discontinuations in 2% (4/161) of patients treated with LEQEMBI. Symptoms of infusion-related reactions included fever and flu-like symptoms (chills, generalized aches, feeling shaky, and joint pain), nausea, vomiting, hypotension, hypertension, and oxygen desaturation.
- After the first infusion, 38% of LEQEMBI-treated patients had transient decreased lymphocyte counts to <0.9 x109/L compared to 2% on placebo, and 22% of LEQEMBI-treated patients had transient increased neutrophil counts to >7.9 x109/L compared to 1% on placebo.
- In the event of an infusion-related reaction, the infusion rate may be reduced, or the infusion may be discontinued, and appropriate therapy initiated as clinically indicated. Prophylactic treatment with antihistamines, acetaminophen, nonsteroidal anti-inflammatory drugs, or corticosteroids prior to future infusions may be considered.
ADVERSE REACTIONS
- In Study 201, 15% of LEQEMBI-treated patients, compared to 6% of placebo-treated patients, stopped study treatment because of an adverse reaction. The most common adverse reaction leading to discontinuation of LEQEMBI was infusion-related reactions that led to discontinuation in 2% (4/161) of patients treated with LEQEMBI compared to 1% (2/245) of patients on placebo.
- The most common adverse reactions reported in ≥5% of patients treated with LEQEMBI (N=161) and ≥2% higher than placebo (N=245) in Study 1 were infusion-related reactions (LEQEMBI: 20%; placebo: 3%), headache (LEQEMBI: 14%; placebo: 10%), ARIA-E (LEQEMBI: 10%; placebo: 1%), cough (LEQEMBI: 9%; placebo: 5%), and diarrhea (LEQEMBI: 8%; placebo: 5%).
| Eisai |
Biogen Inc. |
|
Eisai Co., Ltd.
Public Relations Department
Eisai Inc. (U.S.)
Libby Holman
Eisai Europe, Ltd.
(UK, Europe, Australia, New Zealand and Russia)
EMEA Communications Department
|
Natacha Gassenbach
+ 1-857-777-6573
public.affairs@biogen.com |
Investor Contacts:
| Eisai Co., Ltd. |
Biogen Inc. |
Investor Relations Department
|
Mike Hencke
|
Notes to Editors
1. About LEQEMBITM (lecanemab-irmb)
LEQEMBITM (lecanemab-irmb) is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (protofibril) and insoluble forms of amyloid-beta (Aβ). LEQEMBI is indicated for the treatment of Alzheimer’s disease (AD) in the U.S. This indication is approved under accelerated approval based on reduction in Aβ plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
LEQEMBI is the result of a strategic research alliance between Eisai and BioArctic. Eisai has been initiated submission of data for BLA to the National Medical Products Administration (NMPA) of China in December 2022. Eisai plans to file for marketing authorization applications of lecanemab in Japan and Europe by the end of Eisai’s FY2022.
Since July 2020 Eisai’s Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing. Eisai has completed a LEQEMBI subcutaneous bioavailability study, and subcutaneous dosing is currently being evaluated in the Clarity AD (Study 301) OLE.
2. About Amyloid-Related Imaging Abnormalities (ARIA)
ARIA is an important adverse event of amyloid-lowering therapies that is critical to monitor and manage during treatment. ARIA is most commonly seen as temporary swelling/effusion (ARIA-E) in areas of the brain that usually resolves over time. Some people may also have small spots of bleeding in or on the surface of the brain (ARIA-H) with the swelling. Although most people with ARIA-E do not have symptoms, some people may have symptoms such as headache, confusion, dizziness, vision changes and nausea.
3. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of LEQEMBI development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
4. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market LEQEMBI for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody LEQEMBI back-up was signed in May 2015.
5. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), by working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs.
6. About Biogen
As pioneers in neuroscience, Biogen discovers, develops and delivers worldwide innovative therapies for people living with serious neurological diseases as well as related therapeutic adjacencies. One of the world’s first global biotechnology companies, Biogen was founded in 1978 by Charles Weissmann, Heinz Schaller, Sir Kenneth Murray, and Nobel Prize winners Walter Gilbert and Phillip Sharp. Today, Biogen has a leading portfolio of medicines to treat multiple sclerosis, has introduced the first approved treatment for spinal muscular atrophy, and developed the first approved treatment to address a defining pathology of Alzheimer’s disease. Biogen is also commercializing biosimilars and focusing on advancing one of the industry’s most diversified pipelines in neuroscience that will transform the standard of care for patients in several areas of high unmet need.
The company routinely posts information that may be important to investors on its website at www.biogen.com. To learn more, please visit www.biogen.com and Follow Biogen on social media – Twitter, LinkedIn, Facebook, YouTube.
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements are based on Biogen’s current beliefs and expectations and speak only as of the date of this news release. Biogen does not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
References
1https://www.alzforum.org/news/conference-coverage/lecanemab-sweeps-toxic-av-protofibrils-catches-eyes-trialists
2Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, Lannfelt L, Pettersson FE. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7(2):e32014. doi: 10.1371/journal.pone.0032014. Epub 2012 Feb 15. PMID: 22355408; PMCID: PMC3280222.
Full text(PDF)(243KB)
by codm | Dec 28, 2022 | Newsletter
For Print (PDF)
Yasuda Logistics Corporation (Headquarters: Tokyo, CEO: Nobuyuki Fujii, “Yasuda Logistics”) and Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) announced today that they have entered into a share purchase agreement concerning the transfer of all shares of Eisai’s wholly-owned subsidiary Eisai Distribution Co., Ltd. (Headquarters: Kanagawa, “Eisai Distribution”) to Yasuda Logistics. The date of the share transfer is scheduled to be March 31, 2023.
Eisai Distribution was established in April 1991 as a wholly-owned subsidiary of Eisai and is mainly responsible for logistics-related operations for Eisai Group products, contributing to their stable supply, and has a wealth of experience and know-how specialized in pharmaceutical logistics with a large track record in handling third-party products.
Yasuda Logistics has formulated “Long-term Vision for 2030” that designs what the company should be in 2030, and in order to realize the long-term vision, Yasuda has formulated a Middle-term Management Plan “YASDA Next Challenge 2024”, aiming to build a business structure toward further growth. Yasuda Logistic Group believes that the integration of Eisai Distribution’s prescription drug delivery network with the company’s experience in warehouse operations will improve its medical logistics services, which is an important pillar of its growth strategy, as well as expanding the number of warehouses and other facilities, leading to the realization of higher quality medical logstics services than ever.
Eisai has been working on creating pharmaceutical products and solutions to contribute to patients and the people in the daily living domain with the vision of “empowering people to realize their fullest life.” With the business environment surrounding pharmaceuticals logistics undergoing significant changes, through this agreement Eisai will pursue a more stable and efficient supply of Eisai Group products and concentrate on select business areas. Eisai expects that the synergistic effect of Eisai Distribution entering into the Yasuda Logistics Group will lead to the company’s sustainable growth and further expansion, as well as even greater contributions to the stable supply of pharmaceutical products in Japan.
Eisai Distribution will continue to be responsible for logistics-related operations for Eisai Group products after April 1, 2023. A certain period of time will be set as the transition period, during which Eisai will provide Yasuda Logistics with the necessary support for continuation of Eisai Distribution’s business.
As a result of this transaction, Eisai anticipates no changes to its consolidated financial forecast for the period ended March 31, 2023. Yasuda Logistics also anticipates no changes to its consolidated financial forecast for the period ended March 31, 2023.
Media Inquiries
[Notes to editors]
1. About Eisai Distribution
| Company name: |
Eisai Distribution Co., Ltd. |
| Location: |
1-16-2 Iiyama Minami, Atsugi, Kanagawa |
| Representative: |
Masayuki Miyajima, Representative Director and President |
| Scope of business: |
Distribution (warehousing, storage, delivery) of pharmaceuticals, diagnostics, investigational drugs, pharmaceutical raw materials, foods and food additives, etc., pharmaceutical manufacturing (packaging, labeling, storage), wholesale distribution, warehousing, others |
| Capital: |
60 million JPY |
| Date established: |
April 1, 1991 |
| Number of employees: |
65 (as of March 31, 2022) |
2. About Yasuda Logistics Corporation
Since the establishment in 1919, Yasuda Logistics Corporation has grown its logistics business in scope from the Tokyo metropolitan area to encompass the whole of Japan, and strive to better meet the needs of customers through providing logistics services. Yasuda also directed its energy towards the real estate business early on, continuing to work towards its dream of a wealthier society through the activities of both businesses, a dream that is coming to fruition today.
In order to build a business structure for further growth, the Yasuda Logistics Group has formulated “Long-term Vision for 2030” that designs what we should be in 2030, states that “Through a fusion of the world-renowned YASDA brand and innovative technology, our goal is to become a group that surpasses the expectations of all stakeholders.”
In addition, Yasuda has identified “expansion and improvement of logistics centers, and strengthening service provision systems in medical logistics business” as key strategy of its Middle-term Management Plan for FY 2022 to FY 2024 “YASDA Next Challenge 2024”, and has positioned strengthening pharmaceutical logistics business as a key pillar of Yasuda’s growth strategy.
For more information about Yasuda, please visit https://www.yasuda-soko.co.jp .
3. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), Eisai aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, Eisai strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, Eisai demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), with working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs
by codm | Dec 23, 2022 | Newsletter
For Print (PDF)
TOKYO and CAMBRIDGE, Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, “Eisai”) and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, “Biogen”) announced today that Eisai has initiated submission of data for Biologics License Application (BLA) to the National Medical Products Administration (NMPA) of China for lecanemab (development code: BAN2401), an investigational anti-amyloid beta (Aβ) protofibril antibody.
The registration category of lecanemab was designated as a Category 1 drug (innovative biologics not approved in China or any other countries).
The data submitted in this package includes data from the Phase II clinical trial (Study 201) in mild cognitive impairment (MCI) due to Alzheimer’s disease (AD) and mild AD (collectively known as early AD) with confirmed Aβ accumulation in the brain and the top-line data of the large global Phase III Clarity AD study. Eisai will submit additional data including full data of the Clarity AD study, as directed by the NMPA.
Lecanemab selectively binds and eliminates soluble, toxic Aβ aggregates (protofibrils) that are thought to contribute to the neurotoxicity in AD. As such, lecanemab may have the potential to have an effect on disease pathology and to slow down the progression of the disease. The Clarity AD study of lecanemab met its primary endpoint and all key secondary endpoints with highly statistically significant results. In November 2022, the results of the Clarity AD study were presented at the 2022 Clinical Trials on Alzheimer’s Disease (CTAD) conference, and simultaneously published in the New England Journal of Medicine, peer-reviewed medical journals.
In the U.S., lecanemab was granted Breakthrough Therapy and Fast Track designations by the U.S. Food and Drug Administration (FDA) in June and December 2021, respectively. In July 2022, the FDA accepted Eisai’s BLA for lecanemab under the accelerated approval pathway and granted it Priority Review. The Prescription Drug User Fee Act (PDUFA) action date is January 6, 2023. Eisai aims to file for traditional approval in the U.S. and for marketing authorization applications in Japan and the Europe by the end of Eisai’s FY2022, which ends March 31, 2023.
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
Contacts:
| Eisai Co., Ltd. |
Biogen Inc. |
|
MEDIA CONTACT:
Public Relations Department
TEL: +81-(0)3-3817-5120
INVESTOR CONTACT:
Investor Relations Department
TEL: +81-(0)3-3817-5122
|
MEDIA CONTACT:
Natacha Gassenbach
+ 1-857-777-6573
public.affairs@biogen.com
INVESTOR CONTACT:
Mike Hencke
+ 1-781-464-2442
IR@biogen.com
|
[Notes to editors]
1. About Lecanemab
Lecanemab is an investigational humanized monoclonal antibody for AD that is the result of a strategic research alliance between Eisai and BioArctic. Lecanemab selectively binds and eliminates soluble, toxic amyloid-beta (Aβ) aggregates (protofibrils) that are thought to contribute to the neurotoxicity in AD. As such, lecanemab may have the potential to have an effect on disease pathology and to slow down the progression of the disease. Currently, lecanemab is being developed as the only anti- Aβ antibody that can be used for the treatment of early AD without the need for titration.
Since July 2020 the Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing.
Furthermore, Eisai has initiated a lecanemab subcutaneous dosing Phase 1 study.
2. About Phase II (Study 201) study and Phase III Clarity AD study
Phase II clinical study (Study 201) was conducted as a double-blind, parallel-group, dose-finding study of lecanemab or placebo for 18 months in 856 people living with early AD. Lecanemab treatment resulted in a dose-dependent, longitudinal, and significant reduction in PET SUVR, which assesses amyloid-β accumulation in the brain, compared to placebo. At 18 months, ADCOMS1, CDR-SB2, and ADAS-cog143 showed a dose-dependent reducing clinical decline, with suppression rates of 29.7%, 26.5%, and 47.2% in the 10 mg/kg bi-weekly treatment, respectively. The study did not achieve its primary outcome measure4 at 12 months of treatment. The most common adverse events occurring in the 10 mg/kg biweekly group (incidence ≥ 5% and more frequent than in the placebo group) were infusion reactions (19.9%), headache (13.7%), ARIA-E (9.9%), cough (8.7%), diarrhea (8.1%), dizziness (7.5%), microhemorrhages (5.6%).
1 ADCOMS is developed by Eisai, ADCOMS combines items from the ADAS-Cog scale for assessing cognitive functions, MMSE and the CDR scale for evaluating the severity of dementia to enable highly sensitive detection of changes in clinical functions of early AD symptoms and changes in memory
2 CDR-SB is a numeric scale used to quantify the various severity of symptoms of dementia. Based on interviews of people living with AD and family/caregivers, qualified healthcare professionals assess cognitive and functional performance in six areas: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. The total score of the six areas is the score of CDR-SB, and CDR-SB is also used as an appropriate item for evaluating the effectiveness of therapeutic drugs targeting the early stages of AD.
3 ADAS-Cog is the most common cognitive assessment instrument used in AD clinical trials all over the world. ADAS-Cog14 consists of 14 competencies: word recall, commands, constructional praxis, object and finger naming, ideational praxis, orientation, word recognition, remembering word recognition instructions, comprehension of spoken language, word finding difficulty, spoken language ability, delayed word recall, number cancellation, and maze task. ADAS-Cog has been used in clinical trials for earlier stages of AD including MCI.
4 An 80% or higher estimated probability of demonstrating 25% or greater slowing in clinical decline at 12 months treatment measured by ADCOMS from baseline compared to placebo.
Phase III Clarity AD study was conducted as a placebo-controlled, double-blind, parallel-group, randomized study of lecanemab 10 mg/kg or placebo administered bi-weekly for 18 months in 1,795 people living with early AD. Mean change of CDR-SB from baseline at 18 months as the primary endpoint was 1.21 and 1.66 for lecanemab and placebo groups, respectively. Lecanemab treatment resulted in highly statistically significant results, reducing clinical decline on the global cognitive and functional scale, compared with placebo at 18 months by -0.45 (95% Confidence Interval (CI): -0.67, -0.23; P=0.00005), representing a 27% slowing of decline. Starting as early as six months (difference: -0.17 [95% CI: -0.29, -0.05]; P<0.01), and increasing in absolute difference over time across all time points every 3 months, the treatment showed highly statistically significant changes in CDR-SB from baseline compared to placebo (all p-values are less than 0.01).
All key secondary endpoints, amyloid Positron Emission Tomography (PET) using Centiloids, ADAS-Cog14, ADCOMS and ADCS MCI-ADL5, also showed highly statistically significant results compared with placebo (P<0.001).
The most common adverse events (>10%) in the lecanemab group were infusion reactions (lecanemab: 26.4%; placebo: 7.4%), ARIA-H (combined cerebral microhemorrhages, cerebral macrohemorrhages, and superficial siderosis; lecanemab: 17.3%; placebo: 9.0%), ARIA-E (edema/effusion; lecanemab: 12.6%; placebo: 1.7%), headache (lecanemab: 11.1%; placebo: 8.1%), and fall (lecanemab: 10.4%; placebo: 9.6%). Infusion reactions were largely mild-to-moderate (grade 1-2: 96%) and occurred on the first dose (75%).
During the study period, deaths occurred in 0.7% and 0.8% of participants in the lecanemab and placebo groups, respectively and no deaths were related to lecanemab or occurred with amyloid-related imaging abnormalities (ARIA) in 18-month double-blind study period. Serious adverse events were experienced by 14.0% of participants in the lecanemab group and 11.3% of participants in the placebo group. Treatment-emergent adverse events occurred in 88.9% and 81.9% of participants in the lecanemab and placebo groups, respectively. Treatment-emergent adverse events leading to drug withdrawal occurred in 6.9% and 2.9% of participants in the lecanemab and placebo groups, respectively.
Overall, lecanemab’s ARIA incidence profile was within expectations based on the Phase 2 trial results (Study 201). ARIA-E events were largely mild-to-moderate radiographically (91% of those who had ARIA-E), asymptomatic (78% of those who had ARIA-E), occurred within the first 3 months of treatment (71% of those who had ARIA-E) and resolved within 4 months of detection (81% of those who had ARIA-E). Among the 2.8% of lecanemab-treated subjects with symptomatic ARIA-E, the most commonly reported symptoms were headache, visual disturbance, and confusion. The incidence of symptomatic ARIA-H was 0.7% in the lecanemab group and 0.2% in the placebo group. No imbalance was observed in isolated ARIA-H (i.e., ARIA-H in participants who did not also experience ARIA-E) between lecanemab (8.9%) and placebo (7.8%).
5 ADCS MCI-ADL assesses the competence of patients with MCI in activities of daily living (ADLs), based on 24 questions to the patient’s partner about actual recent activities of daily living.
3. About Registration Categories of Biological Agents (Therapeutic Use) in China
The registration categories of biologics (therapeutic use) in China include categories 1 through 3. Category 1 is for innovative biologics that have not been approved in China or in any other countries; Category 2 is for biologics that improve on products approved in China or any other countries, improving safety, efficacy, quality, and having clear therapeutic superiority; and Category 3 is for biologics that have been approved in China or any other countries. Applications under Category 1 are more likely to receive priority review designation, which is expected to shorten the review period.
4. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
5. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
6. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), with working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs.
7. About Biogen
As pioneers in neuroscience, Biogen discovers, develops, and delivers worldwide innovative therapies for people living with serious neurological diseases as well as related therapeutic adjacencies. One of the world’s first global biotechnology companies, Biogen was founded in 1978 by Charles Weissmann, Heinz Schaller, Sir Kenneth Murray, and Nobel Prize winners Walter Gilbert and Phillip Sharp. Today, Biogen has a leading portfolio of medicines to treat multiple sclerosis, has introduced the first approved treatment for spinal muscular atrophy, and developed the first and only approved treatment to address a defining pathology of Alzheimer’s disease. Biogen is also commercializing biosimilars and focusing on advancing one of the industry’s most diversified pipelines in neuroscience that will transform the standard of care for patients in several areas of high unmet need.
The company routinely posts information that may be important to investors on its website at www.biogen.com. Follow Biogen on social media – Twitter, LinkedIn, Facebook, YouTube.
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements are based on Biogen’s current beliefs and expectations and speak only as of the date of this news release. Biogen does not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
by codm | Dec 23, 2022 | Newsletter
For Print (PDF)
by codm | Dec 22, 2022 | Newsletter
For Print (PDF)
TOKYO and Osaka, December 22, 2022 – Astellas Pharma Inc. (TSE: 4503, President and CEO: Kenji Yasukawa, “Astellas”), Eisai Co., Ltd. (TSE: 4523, CEO: Haruo Naito, “Eisai”), Daiichi Sankyo Company, Limited (TSE: 4568, President : Sunao Manabe, “Daiichi Sankyo”) and Takeda Pharmaceutical Company Limited. (TSE: 4502 / NYSE:TAK, President and CEO Christophe Weber, “Takeda”) today announced that the four companies have agreed the collaboration to reduce environmental burden in the field of pharmaceutical packaging.
Based on the agreement, the four companies will aim to promote the use of more environmentaly friendly packaging for pharmaceutical products by sharing knowledge on packaging technologies to reduce environmental burden, such as blister packs made of biomass-based plastic instead of petroleum-derived plastic, compact packaging, recycled packaging materials, and recyclable packaging materials.
Astellas, Eisai, Daiichi Sankyo, and Takeda aim to ensure that society benefits from this collaboration to harmonize corporate activities with the global environment. In the future, the four companies expect to expand this collaboration beyond the four companies by calling on other companies in order to reduce further environmental burden.
■ Initiatives for Sustainability (Environment) of Each Company
Astellas has set “Deepen our engagement in sustainability” as one of the strategic goals in its Corporate Strategic Plan 2021. The reduction of environmental burden is one of Astellas’ priority themes within sustainability. For more information on specific initiatives, please visit our website.
Eisai established the “Eisai Environmental Management Vision” this fiscal year, and in addition to climate change countermeasures aimed at achieving carbon neutrality by fiscal 2040, Eisai has formed a medium- to long-term plan for environmental issues including efficient use of water and recycling of resources, and will work to further advance these efforts. To learn more about our environmental initiatives, please visit our website.
As a healthcare company with the Purpose “to contribute to the enrichment of quality of life around the world,” Daiichi Sankyo considers global environmental conservation, which is the basis of life and livelihood, as a key management issue (Materiality) and promotes environmental management. For more information on specific initiatives, please visit our website.
At Takeda, “Purpose-led Sustainability” is about creating both business and societal value through its core business. Takeda continues to reduce our operational carbon footprint and are now committed to achieving net-zero GHG emissions for scopes 1 and 2 before 2035 and for Scope 3 before 2040. For more information, see our Annual Integrated Report.
Astellas Pharma Inc.
Corporate Advocacy & Relations
+81 3 3244 3201
Eisai Co., Ltd.
Public Relations Department
+81 3 3817 5120
Daiichi Sankyo Co., Ltd.
Corporate Communications Department
+81 3 6225 1126
Takeda Pharmaceutical Company Limited
Global External Corporate Communications
+81 3 3278 2314






